Ingrediente active: Lenalidomidă
Revlimid 2,5 mg capsule
Revlimid 5 mg capsule
Revlimid 7,5 mg capsule
Revlimid 10 mg capsule
Revlimid 15 mg capsule
Revlimid 20 mg capsule
Revlimid 25 mg capsule
De ce se utilizează Revlimid? Pentru ce este?
Revlimid conține substanța activă "lenalidomidă". Acest medicament aparține unui grup de medicamente care afectează funcționarea sistemului imunitar.
Revlimid este utilizat la adulți pentru:
- Mielom multiplu
- Sindroame mielodisplazice
- Limfom cu celule de manta
Mielom multiplu și Revlimid
Mielomul multiplu este un tip de cancer care afectează un anumit tip de celule albe din sânge, numite plasmocite. Aceste celule se colectează în măduva osoasă și se divid necontrolat. Acest lucru poate deteriora oasele și rinichii.
Mielomul multiplu este de obicei incurabil. Cu toate acestea, semnele și simptomele pot fi reduse sau pot dispărea mult timp. Acest rezultat se numește „răspuns”.
În tratamentul mielomului multiplu, Revlimid este utilizat în asociere cu alte medicamente.
Revlimid la pacienții cu mielom multiplu nou diagnosticat
Revlimid este utilizat numai la pacienții nou diagnosticați atunci când nu pot face un transplant de măduvă osoasă.
Dacă aveți 75 de ani sau mai mult sau aveți probleme renale moderate până la severe, medicul dumneavoastră vă va verifica cu atenție înainte de a începe tratamentul.
Există două tipuri de tratament la pacienții nou diagnosticați:
- Revlimid împreună cu un medicament antiinflamator numit „dexametazonă”.
- Revlimid împreună cu un medicament pentru chimioterapie numit „melphalan” și un medicament imunosupresor numit „prednison”. Veți lua aceste alte medicamente la începutul tratamentului și apoi veți continua să luați Revlimid singur.
Revlimid la pacienții cu mielom multiplu care au avut cel puțin un alt tip de tratament înainte
- Revlimid se administrează împreună cu un medicament antiinflamator numit „dexametazonă”.
Revlimid poate opri agravarea semnelor și simptomelor mielomului multiplu. De asemenea, s-a demonstrat că întârzie revenirea mielomului multiplu după tratament.
Sindroame mielodisplazice și Revlimid
Sindroamele mielodisplazice (MDS) sunt o colecție de multe boli diferite ale sângelui și măduvei osoase. Celulele sanguine devin anormale și nu funcționează corect. Pacienții pot avea o varietate de semne și simptome, inclusiv un număr scăzut de celule roșii din sânge (anemie), o nevoie de transfuzii de sânge și un risc de infecție.
Revlimid pe cont propriu este utilizat pentru tratarea pacienților adulți diagnosticați cu sindroame mielodisplazice care au toate condițiile următoare:
- dacă aveți nevoie de transfuzii regulate de sânge pentru a trata niveluri scăzute de celule roșii din sânge („anemie dependentă de transfuzie”)
- dacă aveți o „anomalie a celulelor măduvei osoase numită„ anomalie citogenetică de deleție 5q izolată. ”Aceasta înseamnă că corpul dumneavoastră nu produce suficiente celule sanguine sănătoase
- dacă alte tratamente utilizate anterior nu sunt adecvate sau nu sunt suficient de eficiente.
Revlimid poate crește numărul de globule roșii sănătoase produse de organism prin reducerea numărului de celule anormale:
- Acest lucru poate reduce numărul de transfuzii de sânge necesare. Nu poate fi necesară nicio transfuzie.
Limfom cu celule de manta si Revlimid
Limfomul cu celule de manta este un cancer al țesutului limfatic (parte a sistemului imunitar), care afectează un tip de celule albe din sânge numite limfocite B. În limfomul cu celule de manta, limfocitele B cresc fără control și se acumulează în țesutul limfatic, măduva osoasă. , sau sânge.
Revlimid este utilizat singur pentru a trata pacienții adulți diagnosticați cu limfom celular de manta netratat anterior.
Cum funcționează Revlimid
Revlimid acționează asupra sistemului imunitar al organismului și direct asupra tumorii în mai multe moduri:
- oprind dezvoltarea celulelor canceroase
- oprind creșterea vaselor de sânge care transportă sângele către celulele tumorale
- prin stimularea unei părți a sistemului imunitar să atace celulele canceroase.
Contraindicații Când Revlimid nu trebuie utilizat
Nu luați Revlimid
- Dacă sunteți gravidă sau credeți că sunteți gravidă sau intenționați să rămâneți gravidă, deoarece se așteaptă ca Revlimid să fie dăunător pentru un copil nenăscut (vezi secțiunea 2, „Avertismente și precauții” și „Sarcina și alăptarea”).
- Dacă există posibilitatea să rămâneți gravidă, cu excepția cazului în care luați toate măsurile necesare pentru a evita sarcina (vezi secțiunea 2 „Avertismente și precauții” și „Sarcina și alăptarea”). Dacă există șansa să rămâneți gravidă, medicul dumneavoastră va nota și va confirma cu fiecare rețetă că au fost luate măsurile necesare pentru a evita sarcina.
- Dacă sunteți alergic la lenalidomidă sau la oricare dintre celelalte componente ale acestui medicament (enumerate la pct. 6). Dacă credeți că sunteți alergic, cereți sfatul medicului dumneavoastră.
Dacă oricare dintre acestea se aplică dumneavoastră, nu luați Revlimid. Dacă aveți dubii, consultați-vă medicul.
Precauții pentru utilizare Ce trebuie să știți înainte de a lua Revlimid
Spuneți medicului dumneavoastră înainte de a începe tratamentul dacă:
- ați avut episoade de cheaguri de sânge în trecut, deoarece riscul formării cheagurilor de sânge în vene și artere crește în timpul tratamentului
- aveți semne de infecție, cum ar fi tuse sau febră
- aveți probleme cu rinichii - medicul dumneavoastră poate modifica doza de Revlimid
- ați avut un atac de cord, ați avut un cheag de sânge sau dacă fumați, aveți tensiune arterială crescută sau niveluri ridicate de colesterol
- are o sarcină tumorală ridicată pe tot corpul, inclusiv în măduva osoasă. Aceasta ar putea duce la o boală în care tumorile se descompun și pot provoca niveluri neobișnuite de substanțe chimice în sânge, care pot duce la insuficiență renală (această boală apare numită „tumoare sindrom de liză ")
- ați avut o reacție alergică în timpul tratamentului cu talidomidă, cum ar fi erupții cutanate, mâncărime, umflături, amețeli sau dificultăți de respirație
Dacă oricare dintre acestea se aplică dumneavoastră, spuneți medicului dumneavoastră înainte de a începe tratamentul.
Dacă aveți sindroame mielodisplazice, este mai probabil să dezvoltați o boală mai avansată numită leucemie mieloidă acută (LMA). În plus, rolul Revlimid asupra probabilității de a dezvolta LMA nu este cunoscut. Medicul dumneavoastră vă poate cere unele teste pentru a verifica semnele care ar putea prezice mai exact probabilitatea de a dezvolta LMA în timpul tratamentului cu Revlimid.
Analize și controale
Veți face regulat teste de sânge înainte și în timpul tratamentului cu Revlimid, deoarece Revlimid poate determina o reducere a celulelor sanguine care vă protejează împotriva infecțiilor (globule albe din sânge) și a celor care ajută la coagularea sângelui (trombocite). Medicul dumneavoastră vă va cere să faceți un test de sânge:
- înainte de tratament
- în fiecare săptămână pentru primele 8 săptămâni de tratament (pentru pacienții cu limfom cu celule de manta, acest lucru se va întâmpla la fiecare 2 săptămâni în ciclurile 3 și 4 și apoi la începutul fiecărui ciclu)
- cel puțin o dată pe lună după aceea.
Medicul dumneavoastră poate verifica modificările pielii, cum ar fi pete roșii sau erupții cutanate.
Medicul dumneavoastră poate decide să ajusteze doza de Revlimid sau să oprească tratamentul în funcție de rezultatele analizelor de sânge și de starea dumneavoastră generală. Dacă sunteți un pacient nou diagnosticat, medicul dumneavoastră poate evalua, de asemenea, tratamentul în funcție de vârsta dumneavoastră și de orice alte afecțiuni care ar putea fi deja prezente.
Donare de sange
Nu trebuie să donați sânge în timpul tratamentului cu lenalidomidă și timp de o săptămână după întreruperea tratamentului.
Copii și adolescenți
Revlimid nu este recomandat pentru utilizare la copii și adolescenți cu vârsta sub 18 ani.
Interacțiuni Ce medicamente sau alimente pot modifica efectul Revlimid
Spuneți medicului dumneavoastră sau asistentei medicale dacă luați, ați luat recent sau s-ar putea să luați orice alte medicamente, inclusiv cele obținute fără prescripție medicală și medicamente pe bază de plante. Acest lucru se datorează faptului că Revlimid poate afecta modul în care acționează alte medicamente modul în care funcționează Revlimid.
Spuneți în special medicului dumneavoastră sau asistentei medicale dacă luați oricare dintre următoarele medicamente:
- unele medicamente utilizate pentru prevenirea sarcinii, cum ar fi contraceptivele orale, deoarece acestea nu mai pot fi eficiente
- unele medicamente utilizate pentru probleme cardiace, cum ar fi digoxina
- unele medicamente utilizate pentru subțierea sângelui, cum ar fi warfarina
Avertismente Este important să știm că:
Sarcina, alăptarea și contracepția - informații pentru femei și bărbați
Sarcina
Pentru femeile care iau Revlimid
- Nu trebuie să luați Revlimid dacă sunteți gravidă, deoarece este de așteptat ca acest medicament să fie dăunător pentru un copil nenăscut.
- Nu trebuie să rămâneți gravidă în timpul tratamentului cu Revlimid. Dacă există posibilitatea unei sarcini, trebuie să utilizați metode contraceptive eficiente (vezi secțiunea „Contracepție”).
- Dacă rămâneți gravidă în timp ce luați Revlimid, trebuie să întrerupeți imediat tratamentul și să informați medicul dumneavoastră.
Pentru bărbații care iau Revlimid
- Dacă partenerul dvs. rămâne gravidă în timp ce luați Revlimid, spuneți imediat medicului dumneavoastră. De asemenea, se recomandă ca partenerul dumneavoastră să vă contacteze medicul.
- În plus, trebuie să utilizați metode eficiente de contracepție (a se vedea secțiunea „Contracepție”).
Timp de hrănire
Nu trebuie să alăptați în timp ce luați Revlimid, deoarece nu se știe dacă acest medicament trece în laptele matern.
Contracepție
Pentru femeile care iau Revlimid
Înainte de a începe tratamentul, trebuie să vă întrebați medicul dacă există posibilitatea să rămâneți gravidă, chiar dacă credeți că este puțin probabil.
Dacă există posibilitatea să rămâneți gravidă
- va trebui să faceți teste de sarcină sub supravegherea medicului dumneavoastră (înainte de fiecare tratament, la fiecare 4 săptămâni în timpul tratamentului și la 4 săptămâni după terminarea tratamentului), cu excepția cazurilor în care s-a confirmat că trompele uterine au fost tăiate și închise, pentru a preveni ouăle ajung la uter (sterilizarea prin legarea tuburilor)
- trebuie să utilizați metode contraceptive eficiente timp de 4 săptămâni înainte de începerea tratamentului, în timpul tratamentului și până la 4 săptămâni după oprirea tratamentului. Medicul dumneavoastră vă va sfătui cu privire la metodele adecvate de contracepție.
Pentru bărbații care iau Revlimid
Revlimid trece în sperma umană. Dacă există posibilitatea ca partenerul dvs. să fie gravidă sau să rămână însărcinată și nu utilizează metode contraceptive eficiente, ar trebui să utilizați prezervative în timpul tratamentului și timp de o săptămână după terminarea tratamentului, chiar dacă ați avut o vasectomie.
Conducerea vehiculelor și utilizarea utilajelor
Nu conduceți vehicule și nu folosiți utilaje dacă vă simțiți amețit, obosit, somnoros, amețit sau aveți vedere încețoșată.
Revlimid conține lactoză
Revlimid conține lactoză. Dacă medicul dumneavoastră v-a spus că aveți o „intoleranță la unele zaharuri, contactați medicul dumneavoastră înainte de a lua Revlimid.
Doză, metodă și timp de administrare Cum se utilizează Revlimid: Doze
Revlimid trebuie administrat de personalul medical care are experiență în tratamentul mielomului multiplu sau a sindroamelor mielodisplazice și a limfomului cu celule de manta.
- Când este utilizat pentru tratamentul mielomului multiplu, Revlimid se administrează în asociere cu alte medicamente (vezi pct. 1 „Ce este„ Revlimid și pentru ce se utilizează ”).
- Când este utilizat pentru tratamentul sindroamelor mielodisplazice și a limfomului cu celule de manta, Revlimid se administrează singur.
Luați întotdeauna Revlimid singur sau Revlimid în asociere cu alte medicamente, urmând întotdeauna exact instrucțiunile medicului. Dacă aveți dubii, consultați medicul sau farmacistul.
Dacă luați Revlimid în asociere cu alte medicamente, vă rugăm să consultați prospectele acestor medicamente pentru mai multe informații despre utilizarea și efectele acestora.
Ciclul de tratament
- Revlimid și medicamentele pe care trebuie să le luați în asociere cu Revlimid se iau în câteva zile pe o perioadă de 4 săptămâni (28 de zile).
- Fiecare perioadă de 28 de zile se numește „ciclu de tratament”.
- În funcție de ziua menstruației, veți lua unul sau mai multe dintre medicamente. Cu toate acestea, în unele zile nu veți lua niciun medicament.
- După finalizarea fiecărui ciclu de 28 de zile, trebuie să înceapă un nou „ciclu” în următoarele 28 de zile.
Doza de Revlimid de luat
Înainte de a începe tratamentul, medicul dumneavoastră vă va spune:
- doza de Revlimid de luat
- doza altor medicamente care trebuie administrată în asociere cu Revlimid, dacă este prescrisă
- în ce zile ale ciclului de tratament să luați fiecare medicament.
Medicul poate observa modificări ale pielii, cum ar fi pete roșii sau erupții cutanate.
Medicul dumneavoastră poate decide, de asemenea, să schimbe doza de Revlimid sau alte medicamente în timpul tratamentului, pe baza rezultatelor analizelor de sânge și a stării dumneavoastră generale (vezi secțiunea 2, „Ce trebuie să știți înainte să luați Revlimid”).
Cum și când să luați Revlimid
- Capsulele trebuie înghițite întregi, de preferință cu apă.
- Nu rupeți, nu deschideți sau mestecați capsulele.
- Capsulele pot fi luate cu sau fără alimente.
- Trebuie să luați Revlimid în zilele stabilite aproximativ la aceeași oră.
Pentru a scoate capsula din blister, apăsați doar pe o parte a capsulei, împingând-o prin folia de aluminiu. Nu apăsați pe centrul capsulei, pentru că se poate rupe.
Durata tratamentului cu Revlimid
Revlimid se administrează în cicluri de tratament, fiecare durând 28 de zile (vezi mai sus „Ciclul de tratament”). Trebuie să continuați cursurile de tratament până când medicul dumneavoastră vă spune să opriți tratamentul.
Dacă uitați să luați Revlimid
Dacă uitați să luați Revlimid la ora obișnuită e
- au trecut mai puțin de 12 ore: luați imediat capsula
- au trecut mai mult de 12 ore: nu luați capsula uitată, ci luați următoarea capsulă a doua zi la ora obișnuită.
Dacă aveți orice întrebări suplimentare cu privire la acest medicament, adresați-vă medicului dumneavoastră sau farmacistului.
Supradozaj Ce trebuie făcut dacă ați luat prea mult Revlimid
Dacă ați luat mai mult Revlimid decât vi s-a spus, spuneți imediat medicului dumneavoastră.
Efecte secundare Care sunt efectele secundare ale Revlimid
Ca toate medicamentele, acest medicament poate provoca reacții adverse, deși nu apar la toate persoanele.
Reacții adverse grave care pot afecta mai mult de 1 din 10 persoane
Revlimid poate reduce numărul de celule albe din sânge care combat infecțiile și celulele sanguine care favorizează coagularea sângelui (trombocite), ceea ce poate duce la tulburări de sângerare, de exemplu. sângerări nazale și vânătăi. Revlimid poate provoca, de asemenea, cheaguri de sânge în vene (tromboză).
Prin urmare, trebuie să solicitați asistență medicală imediată dacă prezentați oricare dintre următoarele reacții adverse:
- febră, frisoane, dureri în gât, tuse, ulcer la gură sau orice alte simptome de infecție (inclusiv în fluxul sanguin (sepsis))
- sângerări sau vânătăi în absența rănilor
- durere în piept sau picioare
- dificultăți de respirație.
Dacă aveți oricare dintre reacțiile adverse enumerate mai sus, vă rugăm să spuneți imediat medicului dumneavoastră.
Alte reacții adverse sunt enumerate mai jos
Este important de reținut că un număr mic de pacienți pot dezvolta alte tipuri de cancer și este posibil ca acest risc să crească odată cu tratamentul cu Revlimid; prin urmare, medicul dumneavoastră trebuie să cântărească cu atenție beneficiul și riscul atunci când vă prescrie Revlimid.
Reacțiile adverse foarte frecvente pot afecta mai mult de 1 din 10 persoane:
- Scăderea numărului de celule roșii din sânge (anemie), care poate provoca oboseală și slăbiciune
- Constipație, diaree, greață, roșeață a pielii, erupții cutanate, vărsături, crampe musculare, dureri musculare, dureri osoase, dureri articulare, oboseală, umflături generalizate, inclusiv umflarea brațelor și picioarelor
- Simptome de febră și gripă, inclusiv febră, dureri musculare, cefalee, dureri de urechi și frisoane
- Amorțeală, furnicături sau senzație de arsură pe piele, durere la nivelul mâinilor sau picioarelor, amețeli, tremor, modificări ale gustului
- Durere toracică iradiată către brațe, gât, maxilar, spate sau stomac, cu senzație de transpirație și dificultăți de respirație, greață sau vărsături, care pot fi simptome ale unui atac de cord (infarct miocardic)
- Reducerea poftei de mâncare
- Niveluri scăzute de potasiu în sânge
- Durere la picioare (care poate fi un simptom al trombozei), durere în piept sau dificultăți de respirație (care pot fi simptome ale cheagurilor de sânge în plămâni, numite embolie pulmonară)
- Infecții de orice fel
- Infecția plămânilor și a căilor respiratorii superioare, dificultăți de respirație
- Vedere încețoșată
- Vedere încețoșată (cataractă)
- Probleme la rinichi
- Modificări ale unei proteine din sânge care pot provoca umflarea arterelor (vasculită)
- Creșteri ale zahărului din sânge (diabet)
- Durere de cap
- Piele uscata
- Dureri de stomac
- Schimbarea dispoziției, dificultăți de somn
Reacțiile adverse frecvente pot afecta până la 1 din 10 persoane:
- Infecția sinusurilor care înconjoară nasul
- Sângerări de la nivelul gingiilor, stomacului sau intestinelor
- Durere crescută, dimensiunea tumorii, roșeață în jurul tumorii
- Creșterea sau scăderea tensiunii arteriale, bătăile lente, rapide sau neregulate ale inimii
- Întunecarea pielii
- Erupții cutanate, crăparea pielii, descuamarea sau descuamarea
- Urticarie, mâncărime, transpirație crescută, deshidratare
- Gură dureroasă cu ulcer, gură uscată, dificultate la înghițire
- Dureri de stomac
- Producția de urină mult mai mult sau mai puțin decât de obicei (care poate fi un simptom al insuficienței renale), sânge în urină
- Dificultăți de respirație, în special când stați culcat (care ar putea fi un simptom al insuficienței cardiace)
- Dificultăți de a obține o erecție
- Accident vascular cerebral, leșin
- Slabiciune musculara
- Umflarea articulațiilor
- Modificări ale hormonului tiroidian din sânge, niveluri scăzute de calciu, fosfat sau magneziu din sânge
- Depresie
- Surditate
- Teste anormale ale funcției hepatice
- Tulburări de echilibru, dificultăți de mișcare
- Sunete în urechi (tinitus)
- Suprasolicitare de fier
- Sete
- Confuzie
- Durere de dinţi
- Pierdere în greutate.
Reacțiile adverse mai puțin frecvente pot afecta până la 1 din 100 de persoane:
- Hemoragie în interiorul craniului
- Probleme circulatorii
- Pierderea vederii
- Pierderea apetitului sexual (libidoul)
- Flux de urină abundent cu durere și slăbiciune a oaselor, care ar putea fi simptome ale unei tulburări renale (sindrom Fanconi)
- Dureri de stomac, balonare sau diaree, care pot fi simptome ale inflamației intestinului gros (numită colită sau tiflită)
- Producerea mult mai mult sau mai puțin de urină decât de obicei, care poate fi un simptom al unui tip de problemă renală (numită necroză tubulară renală)
- Decolorarea pielii, sensibilitate la lumina soarelui
- Unele tipuri de cancer de piele
- Urticarie, erupții cutanate, umflarea ochilor, gurii sau feței, dificultăți de respirație sau mâncărime, care pot fi simptome ale unei reacții alergice.
Reacțiile adverse rare pot afecta până la 1 din 1000 de persoane:
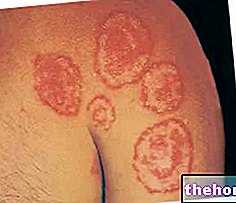
- Reacție alergică severă, care poate începe ca o erupție cutanată într-o zonă, dar se răspândește cu o pierdere extinsă a pielii pe tot corpul (sindrom Stevens-Johnson și / sau necroliză epidermică toxică).
- Sindromul de liză tumorală - complicații metabolice care pot apărea în timpul tratamentului tumorii și uneori chiar și fără tratament. Aceste complicații sunt cauzate de produsele descompuse ale celulelor canceroase pe moarte și pot include următoarele complicații: modificări ale parametrilor hematologici; valori ridicate ale potasiului, fosforului și acidului uric; și valori scăzute ale calciului care duc, în consecință, la modificări ale funcției rinichilor, ritmului cardiac, convulsii și, uneori, moarte.
Frecvență necunoscută: frecvența nu poate fi estimată din datele disponibile:
- Durere bruscă sau ușoară, dar înrăutățită la nivelul abdomenului superior și / sau al spatelui, care persistă câteva zile, posibil cu greață, vărsături, febră și puls rapid.Aceste simptome se pot datora inflamației pancreasului.
- Respirație șuierătoare, dificultăți de respirație sau tuse uscată, care ar putea fi simptome cauzate de inflamația țesutului pulmonar.
- Decolorarea gălbuie a pielii, a mucoaselor sau a ochilor (icter), scaune de culoare deschisă, urină de culoare închisă, mâncărime a pielii, erupții cutanate, durere sau umflarea abdomenului.Acestea ar putea fi simptome de afectare a ficatului (boală hepatică).
- Au fost observate cazuri rare de descompunere musculară (durere, slăbiciune sau umflare a mușchilor), care pot provoca probleme renale (rabdomioliză), dintre care unele când Revlimid este administrat cu o statină (un tip de medicament care scade colesterolul).
- O boală care afectează pielea și este cauzată de inflamația vaselor de sânge mici, cu dureri articulare și febră (vasculită leucocitoclastică).
- Deteriorarea stomacului sau a peretelui intestinal, care poate duce la infecții foarte grave.Spuneți medicului dumneavoastră dacă aveți dureri abdominale severe, febră, greață, vărsături, sânge în scaun sau modificări ale obiceiurilor intestinale.
Raportarea efectelor secundare
Dacă manifestați orice reacții adverse, adresați-vă medicului dumneavoastră, farmacistului sau asistentei medicale.Acestea includ orice posibile reacții adverse nemenționate în acest prospect. De asemenea, puteți raporta reacțiile adverse direct prin intermediul sistemului național de raportare enumerat în Anexa V. Reacțiile adverse pe care le puteți ajuta furnizați mai multe informații despre siguranța acestui medicament.
Expirare și reținere
- Nu lăsați acest medicament la vederea și îndemâna copiilor.
- Nu utilizați acest medicament după data de expirare înscrisă pe blister și cutie după „EXP”. Data de expirare se referă la ultima zi a lunii respective.
- Acest medicament nu necesită condiții speciale de păstrare.
- Nu utilizați acest medicament dacă observați pachete deteriorate sau care prezintă semne de manipulare.
- Nu aruncați niciun medicament prin apele uzate sau deșeurile menajere. Întrebați farmacistul cum să aruncați medicamentele pe care nu le mai utilizați. Acest lucru va ajuta la protejarea mediului.
Termen limită "> Alte informații
Ce conține Revlimid
Revlimid 2,5 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține 2,5 mg lenalidomidă.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină, dioxid de titan (E171), carmin indigo (E132) și oxid galben de fier (E172)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 5 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține lenalidomidă 5 mg.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină și dioxid de titan (E171)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 7,5 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține 7,5 mg de lenalidomidă.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină, dioxid de titan (E171), oxid galben de fier (E172)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 10 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține 10 mg lenalidomidă.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- prepararea capsulelor: gelatină, dioxid de titan (E171), carmin indigo (E132) și oxid de fier galben (E172)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 15 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține 15 mg de lenalidomidă.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină, dioxid de titan (E171) și indigo carmin (E132)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 20 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține lenalidomidă 20 mg.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină, dioxid de titan (E171), carmin indigo (E132) și oxid de fier galben (E172)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Revlimid 25 mg capsule:
- Ingredientul activ este lenalidomida. Fiecare capsulă conține lenalidomidă 25 mg.
- Celelalte ingrediente sunt:
- conținut de capsulă: lactoză anhidră, celuloză microcristalină, croscarmeloză sodică și stearat de magneziu
- învelișul capsulei: gelatină și dioxid de titan (E171)
- cerneală cu litere: șelac, propilen glicol, hidroxid de potasiu și oxid de fier negru (E172).
Cum arată Revlimid și conținutul ambalajului
Capsulele rigide de 2,5 mg Revlimid sunt albastru-verzui / alb, marcate cu „REV 2,5 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând una sau trei blistere. Fiecare blister conține șapte capsule, pentru un total de 7 sau 21 de capsule pe ambalaj.
Capsulele rigide Revlimid 5 mg sunt albe, marcate cu „REV 5 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând una sau trei blistere. Fiecare blister conține șapte capsule, pentru un total de 7 sau 21 de capsule pe ambalaj.
Capsulele rigide Revlimid 7,5 mg sunt de culoare galben pal / alb, marcate cu „REV 7,5 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând trei blistere. Fiecare blister conține șapte capsule, pentru un total de 21 de capsule pe ambalaj.
Capsulele de 10 mg Revlimid sunt albastru-verzui / galben deschis, marcate cu „REV 10 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând trei blistere. Fiecare blister conține șapte capsule, pentru un total de 21 de capsule pe ambalaj.
Capsulele Revlimid 15 mg sunt albastru deschis / alb, marcate cu „REV 15 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând trei blistere. Fiecare blister conține șapte capsule, pentru un total de 21 de capsule pe ambalaj.
Capsulele rigide Revlimid 20 mg sunt albastru-verzui / albastru deschis, marcate cu „REV 20 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând trei blistere. Fiecare blister conține șapte capsule, pentru un total de 21 de capsule pe ambalaj.
Capsulele de 25 mg Revlimid sunt albe, marcate cu „REV 25 mg”.
Capsulele sunt livrate în ambalaje, fiecare conținând trei blistere. Fiecare blister conține șapte capsule, pentru un total de 21 de capsule pe ambalaj.
Prospect sursă: AIFA (Agenția italiană pentru medicamente). Conținut publicat în ianuarie 2016. Este posibil ca informațiile prezente să nu fie actualizate.
Pentru a avea acces la cea mai actualizată versiune, este recomandabil să accesați site-ul web AIFA (Agenția italiană pentru medicamente). Declinare de responsabilitate și informații utile.
01.0 DENUMIREA MEDICAMENTULUI -
REVLIMID 10 MG CAPSULE DURI
▼ Medicament supus unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă a noilor informații de siguranță. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată. Vezi pct. 4.8 pentru informații despre cum să raportezi reacțiile adverse.
02.0 COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ -
Fiecare capsulă conține lenalidomidă 10 mg.
Excipienți cu efecte cunoscute:
Fiecare capsulă conține 294 mg lactoză anhidră.
Pentru lista completă a excipienților, vezi secțiunea 6.1.
03.0 FORMA FARMACEUTICĂ -
Capsula tare.
Capsule albastru-verzi / galben deschis, dimensiune 0,21,7 mm, marcate cu "REV 10 mg".
04.0 INFORMAȚII CLINICE -
04.1 Indicații terapeutice -
Mielom multiplu
Revlimid este indicat pentru tratamentul pacienților adulți cu mielom multiplu netratat anterior care nu sunt eligibili pentru transplant (vezi pct. 4.2).
Revlimid, în combinație cu dexametazona, este indicat pentru tratamentul pacienților adulți cu mielom multiplu care au primit cel puțin o terapie anterioară.
Sindroame mielodisplazice
Revlimid este indicat pentru tratamentul pacienților cu anemie dependentă de transfuzie din cauza sindroamelor mielodisplazice (SMD) cu risc scăzut sau intermediar 1 asociate cu anomalie citogenetică de deleție 5q izolată, atunci când alte opțiuni de tratament sunt insuficiente sau inadecvate.
Limfom cu celule de manta
Revlimid este indicat pentru tratamentul pacienților adulți cu limfom cu celule de manta recidivante sau refractare (vezi pct. 4.4 și 5.1).
04.2 Doze și mod de administrare -
Tratamentul cu Revlimid trebuie supravegheat de un medic cu experiență în utilizarea terapiilor împotriva cancerului (vezi pct. 4.4, cariotip).
Dozare
Mielom multiplu nou diagnosticat
Lenalidomida în asociere cu dexametazonă până la progresia bolii, la non-pacienți eligibil pentru transplant
Tratamentul cu lenalidomidă nu trebuie inițiat dacă numărul absolut de neutrofile (Absolut
Numărul de neutrofile, ANC) este
Doza recomandată
Doza inițială recomandată de lenalidomidă este de 25 mg pe cale orală o dată pe zi, în zilele 1-21 ale ciclurilor repetate de 28 de zile. Doza recomandată de dexametazonă este de 40 mg pe cale orală o dată pe zi, în zilele 1, 8, 15 și 22 ale ciclurilor repetate de 28 de zile. Pacienții pot continua terapia cu lenalidomidă și dexametazonă până la apariția progresiei bolii sau a intoleranței.
Doza poate fi continuată sau modificată pe baza rezultatelor clinice și de laborator (vezi pct. 4.4). Pentru pacienții cu vârsta ≥ 75 de ani, doza inițială de dexametazonă este de 20 mg / zi în zilele 1, 8, 15 și 22 ale fiecărui ciclu de tratament de 28 de zile. Doza recomandată de lenalidomidă la pacienții cu insuficiență renală moderată este de 10 mg o dată pe zi.
Ajustările dozei recomandate în timpul tratamentului și la reluarea tratamentului
După cum se rezumă în tabelele de mai jos, sunt recomandate ajustări ale dozei pentru tratamentul trombocitopeniei și neutropeniei de gradul 3 sau 4 sau pentru gestionarea oricărei alte toxicități de gradul 3 sau 4 despre care se crede că este legată de lenalidomidă.
• Niveluri de reducere a dozei
• Trombocitopenie
a Dacă apare toxicitate limitativă a dozei (Toxicitate care limitează doza, DLT)> Ziua 15 a unui ciclu, dozarea lenalidomidei va fi oprită pentru cel puțin restul ciclului curent de 28 de zile.
• Neutropenie
În caz de neutropenie, medicul trebuie să ia în considerare utilizarea factorilor de creștere în gestionarea pacientului.
Dacă doza de lenalidomidă a fost redusă datorită DLT hematologic, doza de lenalidomidă poate fi reintrodusă la următorul nivel de doză mai mare (până la doza inițială), la discreția medicului curant, dacă tratamentul continuu cu lenalidomidă / dexametazonă a produs măduva osoasă îmbunătățită funcție (absența DLT timp de cel puțin 2 cicluri consecutive și ANC ≥ 1.500 / mcl, cu număr de trombocite ≥ 100.000 / mcl, la începutul unui nou ciclu la doza actuală).
Lenalidomidă în asociere cu melfalan și prednison, urmată de monoterapie de întreținere, în pacienții care nu sunt eligibili pentru transplant
Tratamentul cu lenalidomidă nu trebuie inițiat dacă ANC este
Doza recomandată
Doza inițială recomandată este de lenalidomidă 10 mg / zi pe cale orală în zilele 1-21 din cicluri repetate de 28 de zile timp de până la 9 cicluri, melfalan 0,18 mg / kg pe cale orală în zilele 1-4 din cicluri repetate de 28 de zile, prednison 2 mg / kg oral în zilele 1-4 ale ciclurilor repetate de 28 de zile. Pacienții care finalizează 9 cicluri sau nu pot finaliza terapia combinată din cauza intoleranței trebuie tratați cu monoterapie cu lenalidomidă, 10 mg / zi pe cale orală, în zilele 1-21 de cicluri repetate.28 zile până la progresia bolii. Doza poate fi continuată sau modificată pe baza rezultatelor clinice și de laborator (vezi pct. 4.4).
Ajustările dozei recomandate în timpul tratamentului și la reluarea tratamentului
După cum se rezumă în tabelele de mai jos, sunt recomandate ajustări ale dozei pentru tratamentul trombocitopeniei sau neutropeniei de gradul 3 sau 4 sau pentru gestionarea oricărei alte toxicități de gradul 3 sau 4 despre care se crede că este legată de lenalidomidă.
• Niveluri de reducere a dozei
Dacă neutropenia este singura toxicitate la orice dozare, adăugați factor de stimulare a coloniei de granulocite (G-CSF) și mențineți doza de lenalidomidă..
• Trombocitopenie
• Neutropenie
a Dacă subiectul nu a primit terapie G-CSF, inițiați terapia G-CSF. În Ziua 1 a ciclului următor, continuați GCSF după cum este necesar și mențineți doza de melfalan dacă neutropenia a fost singura DLT. În caz contrar, reduceți un nivel de doză la începutul ciclului următor.
În caz de neutropenie, trebuie luată în considerare utilizarea factorilor de creștere în managementul pacienților.
Mielom multiplu cu cel puțin o terapie anterioară
Doza recomandată
Doza inițială recomandată este de 25 mg lenalidomidă pe cale orală o dată pe zi în zilele 1-21 ale ciclurilor repetate de 28 de zile. Doza recomandată de dexametazonă este de 40 mg pe cale orală o dată pe zi în zilele 1-4, 9-12 și 17-20 din fiecare ciclu de 28 de zile pentru primele 4 cicluri de terapie și 40 mg o dată pe zi după aceea. Zi în zilele 1-4 la fiecare 28 de zile.
Doza poate fi continuată sau modificată pe baza rezultatelor clinice și de laborator (vezi pct. 4.4). Medicii trebuie să evalueze cu atenție doza de dexametazonă de utilizat, luând în considerare starea pacientului și starea bolii.
Tratamentul cu lenalidomidă nu trebuie inițiat dacă ANC este măduva osoasă de către plasmocite, dacă numărul trombocitelor este
Ajustările dozei recomandate în timpul tratamentului și la reluarea tratamentului
După cum se rezumă în tabelele de mai jos, sunt recomandate ajustări ale dozei pentru tratamentul neutropeniei sau trombocitopeniei de gradul 3 sau 4 sau pentru gestionarea oricărei toxicități de gradul 3 sau 4 despre care se crede că este legată de lenalidomidă.
• Niveluri de reducere a dozei
• Trombocitopenie
• Neutropenie
În caz de neutropenie, trebuie luată în considerare utilizarea factorilor de creștere în managementul pacienților.
Sindroame mielodisplazice
Tratamentul cu lenalidomidă nu trebuie inițiat dacă ANC este
Doza recomandată
Doza inițială recomandată este de 10 mg lenalidomidă pe cale orală o dată pe zi, în zilele 1-21 ale ciclurilor repetate de 28 de zile. Doza poate fi continuată sau modificată pe baza rezultatelor clinice și de laborator (vezi pct. 4.4).
Ajustările dozei recomandate în timpul tratamentului și la reluarea tratamentului
După cum se rezumă în tabelele de mai jos, sunt recomandate ajustări ale dozei pentru tratamentul neutropeniei sau trombocitopeniei de gradul 3 sau 4 sau pentru gestionarea oricărei toxicități de gradul 3 sau 4 despre care se crede că este legată de lenalidomidă.
• Niveluri de reducere a dozei
Pentru pacienții care încep cu o doză de 10 mg și care prezintă trombocitopenie sau neutropenie:
• Trombocitopenie
• Neutropenie
Suspendarea lenalidomidei
Pacienții care nu au cel puțin un răspuns eritroid ușor în termen de 4 luni de la începerea tratamentului, demonstrat de o reducere de cel puțin 50% a necesităților de transfuzie sau, dacă nu primesc transfuzii, de o creștere a hemoglobinei cu 1 g / dl, ar trebui să oprească lenalidomida tratament.
Limfom cu celule de manta
Doza recomandată
Doza inițială recomandată este de 25 mg lenalidomidă pe cale orală o dată pe zi în zilele 1-21 ale ciclurilor repetate de 28 de zile.
Testul este continuat sau modificat pe baza constatărilor clinice și de laborator (vezi pct. 4.4).
Ajustările dozei recomandate în timpul tratamentului și la reluarea tratamentului
După cum se rezumă în tabelele de mai jos, sunt recomandate ajustări ale dozei pentru tratamentul neutropeniei sau trombocitopeniei de gradul 3 sau 4 sau pentru gestionarea oricărei toxicități de gradul 3 sau 4 despre care se crede că este legată de lenalidomidă.
• Niveluri de reducere a dozei
1 - În țările în care este disponibilă capsula de 2,5 mg.
• Trombocitopenie
• Neutropenie
• Reacție de erupție tumorală
Tratamentul cu lenalidomidă poate fi continuat la pacienții cu Reacție de erupție tumorală, Indemnizația de concediere de gradul 1 sau 2, fără întrerupere sau modificare, la aprecierea medicului. La pacienții cu TFR de gradul 3 sau 4, tratamentul cu lenalidomidă trebuie întrerupt până când TFR este redus la ≤ gradul 1; Pentru gestionarea simptomelor, pacienții pot fi tratați în conformitate cu liniile directoare TFR de gradul 1 și 2 (vezi pct. 4.4).
Toți pacienții
Pentru alte efecte toxice de gradul 3 sau 4 despre care se crede că sunt legate de lenalidomidă, tratamentul trebuie întrerupt și reluat la următoarea doză mai mică numai atunci când toxicitatea a scăzut la ≤ gradul 2 la discreția medicului.
Întreruperea sau întreruperea tratamentului cu lenalidomidă trebuie luată în considerare în caz de erupție cutanată de gradul 2 sau 3. Tratamentul cu lenalidomidă trebuie întrerupt în caz de angioedem, erupție cutanată de grad 4, exfoliativă sau erupție buloasă sau dacă este suspectat Stevens-Johnson (SSJ) sau epidermic toxic necroliză (NET) și nu trebuie reluată după întreruperea cauzată de aceste reacții.
Populații speciale
Populația pediatrică
Revlimid nu trebuie utilizat la copii și adolescenți de la naștere până la vârsta de 18 ani din motive de siguranță (vezi pct. 4.4).
Pacienți vârstnici
Datele farmacocinetice disponibile în prezent sunt descrise în secțiunea 5.2. Lenalidomida a fost utilizată în studii clinice la pacienți cu mielom multiplu până la vârsta de 91 de ani, la pacienții cu sindroame mielodisplazice cu vârsta de până la 95 de ani și la pacienții cu limfom cu celule de manta până la vârsta de 88 de ani (vezi pct. 5.1).
La pacienții nou diagnosticați cu mielom multiplu cu vârsta de 75 de ani și peste tratați cu lenalidomidă, a existat o incidență mai mare a reacțiilor adverse grave și a reacțiilor adverse care au dus la întreruperea tratamentului (vezi pct. 4.4). trebuie evaluat cu atenție înainte de a lua în considerare tratamentul (vezi pct. 4.4).
• Mielom multiplu nou diagnosticat
Pentru pacienții cu vârsta peste 75 de ani tratați cu lenalidomidă în asociere cu dexametazonă, doza inițială de dexametazonă este de 20 mg / zi în zilele 1, 8, 15 și 22 ale fiecărui ciclu de tratament de 28 de zile.
Nu sunt propuse ajustări ale dozei la pacienții cu vârsta peste 75 de ani tratați cu lenalidomidă în asociere cu melfalan și prednison.
În studiile clinice cu mielom multiplu nou diagnosticat la pacienți neeligibili pentru transplant, terapia combinată cu lenalidomidă a fost mai puțin tolerată la pacienții cu vârsta peste 75 de ani decât la populația mai tânără. Dintre acești pacienți, procentul care a întrerupt tratamentul din cauza intoleranței (evenimente adverse de gradul 3 sau 4 și evenimente adverse grave) a fost mai mare decât la pacienții cu vârsta
• Mielom multiplu tratat anterior cu cel puțin o terapie
Procentul pacienților cu mielom multiplu în vârstă de 65 de ani sau mai mult nu a fost semnificativ diferit între grupurile de lenalidomidă / dexametazonă și placebo / dexametazonă. În general, nu au fost observate diferențe de siguranță și eficacitate între acești pacienți și pacienții mai tineri, deși nu poate fi exclusă o predispoziție mai mare la pacienții vârstnici.
• Sindroame mielodisplazice
Pentru pacienții cu sindroame mielodisplazice tratați cu lenalidomidă, nu s-a observat nicio diferență globală în ceea ce privește siguranța și eficacitatea între pacienții cu vârsta peste 65 de ani și pacienții mai tineri.
• Limfom cu celule de manta
La pacienții cu limfom cu celule de manta tratați cu lenalidomidă, nu s-a observat nicio diferență globală în ceea ce privește siguranța și eficacitatea între pacienții cu vârsta de 65 de ani și peste și pacienții cu vârsta sub 65 de ani.
Deoarece pacienții vârstnici sunt mai predispuși să aibă o funcție renală afectată, ar trebui acordată o atenție deosebită selecției dozei, iar monitorizarea funcției renale trebuie efectuată ca măsură de precauție.
Pacienți cu insuficiență renală
Lenalidomida este substanțial excretată prin rinichi; la pacienții cu grade mai mari de insuficiență renală, tolerabilitatea tratamentului poate fi modificată (vezi pct. 4.4). Trebuie acordată o atenție deosebită alegerii dozelor și se recomandă monitorizarea funcției renale.
Nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară și mielom multiplu, sindroame mielodisplazice sau limfom cu celule de manta. La pacienții cu insuficiență renală moderată sau severă sau cu boală renală în stadiu terminal, la inițierea tratamentului și pe toată durata tratamentului, sunt recomandate următoarele ajustări ale dozei. ) (CLcr dializă).
• Mielom multiplu
1 Doza poate fi crescută la 15 mg o dată pe zi după 2 cicluri dacă pacientul nu răspunde la tratament, dar tolerează medicamentul.
2 În țările în care este disponibilă capsula de 7,5 mg.
• Sindroame mielodisplazice
* Nivelurile recomandate de reducere a dozei în timpul tratamentului și la reluarea tratamentului, pentru a gestiona neutropenia sau trombocitopenia de gradul 3 sau 4 sau alte tipuri de toxicitate de gradul 3 sau 4 considerate legate de lenalidomidă, așa cum este descris mai sus.
• Limfom cu celule de manta
1 Doza poate fi crescută la 15 mg o dată pe zi după 2 cicluri dacă pacientul nu răspunde la tratament, dar tolerează medicamentul.
2 În țările în care este disponibilă capsula de 7,5 mg.
După inițierea terapiei cu lenalidomidă, modificarea ulterioară a dozei de lenalidomidă la pacienții cu insuficiență renală trebuie să se bazeze pe tolerabilitatea tratamentului pentru fiecare pacient, așa cum este descris mai sus.
Pacienți cu insuficiență hepatică
Lenalidomida nu a fost studiată oficial la pacienții cu insuficiență hepatică și nu există recomandări specifice de dozare.
Mod de administrare
Utilizare orală.
Capsulele Revlimid trebuie administrate în zilele desemnate, cam în aceeași oră. Capsulele nu trebuie deschise, sparte sau mestecate. Capsulele trebuie înghițite întregi, de preferință cu apă, cu sau fără alimente. Pacientul poate lua o doză uitată dacă este la mai puțin de 12 ore după ora programată de administrare a acesteia. Dacă, pe de altă parte, este mai mult de 12 ore, pacientul nu trebuie să ia doza uitată, ci să aștepte cantitatea obișnuită. ora următoare pentru a lua următoarea doză.
Se recomandă aplicarea presiunii doar pe o parte a capsulei pentru a o scoate din blister, reducând astfel riscul deformării sau ruperii.
04.3 Contraindicații -
• Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
• Femeile gravide.
• Femeile aflate la vârsta fertilă, cu excepția cazului în care sunt îndeplinite toate condițiile din Programul de prevenire a sarcinii (vezi secțiunile 4.4 și 4.6).
04.4 Avertismente speciale și precauții adecvate pentru utilizare -
Precauții în caz de sarcină
Lenalidomida este legată structural de talidomidă, o substanță activă cu efect teratogen cunoscut la om, care provoacă defecte congenitale grave care pun viața în pericol. Lenalidomida a indus malformații la maimuțe similare celor descrise pentru talidomidă (vezi secțiunile 4.6 și 5.3) .un efect teratogen al lenalidomidei este de așteptat la om în timpul sarcinii.
Condițiile programului de prevenire a sarcinii trebuie îndeplinite pentru toți pacienții, cu excepția cazului în care există dovezi ferme că pacientul nu este capabil să conceapă.
Criterii pentru stabilirea faptului că o femeie nu are un potențial fertil
Un pacient de sex feminin sau partenerul unui pacient de sex masculin este considerat capabil să conceapă, cu excepția cazului în care îndeplinește cel puțin unul dintre următoarele criterii:
• Vârsta ≥ 50 de ani și amenoreea naturală * timp de ≥ 1 an
• Insuficiență ovariană prematură confirmată de un ginecolog
• Salpingo-ooforectomie sau histerectomie bilaterală anterioară
• genotip XY, sindrom Turner, agenezie uterină.
* Amenoreea după terapia anticancerigenă sau în timpul alăptării nu exclude fertilitatea potențială.
Orientare
Lenalidomida este contraindicată femeilor aflate la vârsta fertilă, cu excepția cazului în care sunt îndeplinite toate condițiile următoare:
• Pacientul este conștient de faptul că este de așteptat un risc teratogen pentru făt
• Pacientul este conștient de necesitatea de a utiliza metode contraceptive eficiente, fără întrerupere, cu 4 săptămâni înainte de începerea tratamentului, pe toată durata tratamentului și până la 4 săptămâni după terminarea tratamentului.
• Chiar și în prezența amenoreei, un pacient aflat la vârsta fertilă trebuie să respecte toate recomandările pentru o contracepție eficientă
• Pacientul trebuie să poată respecta măsuri contraceptive eficiente
• Pacienta este informată și conștientă de consecințele potențiale ale sarcinii și de necesitatea de a solicita asistență medicală imediată dacă există riscul de sarcină
• Pacienta este conștientă de necesitatea de a începe tratamentul de îndată ce lenalidomida este eliberată în urma unui test de sarcină negativ
• Pacienta este conștientă de necesitate și este de acord să se supună testelor de sarcină la fiecare 4 săptămâni, cu excepția cazurilor de sterilizare confirmată prin ligatură tubară
• Pacienta recunoaște că este conștientă de riscurile și precauțiile necesare asociate cu utilizarea lenalidomidei
Pentru pacienții de sex masculin care iau lenalidomidă, studiile farmacocinetice au arătat că, în timpul tratamentului, lenalidomida este prezentă la niveluri extrem de scăzute în material seminal și este nedetectabilă în materialul seminal al subiecților sănătoși la 3 zile după oprirea substanței (vezi punctul 5.2). Ca măsură de precauție, toți pacienții de sex masculin care iau lenalidomidă trebuie să îndeplinească următoarele condiții:
• Fiți conștienți de riscul teratogen preconizat în cazul activității sexuale cu o femeie gravidă sau cu potențial fertil.
• Fiți conștienți de necesitatea utilizării prezervativelor în caz de activitate sexuală cu o femeie însărcinată sau potențial fertilă care nu utilizează contracepție eficientă (chiar dacă bărbatul a suferit o vasectomie) în timpul tratamentului și timp de 1 săptămână după suspendarea dozei și / sau tratamentul întreruperea.
• Rețineți că, dacă partenerul rămâne gravidă în timp ce pacientul ia Revlimid sau la scurt timp după întreruperea tratamentului cu Revlimid, acesta trebuie să informeze imediat medicul și să îl trimită pe partener la un specialist sau teratolog care poate evalua situația și poate da o opinie.
În cazul femeilor aflate la vârsta fertilă, medicul trebuie să se asigure că:
• Pacienta îndeplinește cerințele Programului de prevenire a sarcinii, inclusiv confirmarea că are un nivel adecvat de înțelegere
• Pacientul a acceptat condițiile menționate mai sus
Contracepție
Femeile aflate la vârsta fertilă trebuie să utilizeze contracepția eficientă timp de 4 săptămâni înainte de terapie, în timpul terapiei și până la 4 săptămâni după terapia cu lenalidomidă și, de asemenea, în caz de întrerupere a dozei, cu excepția cazului în care pacientul se angajează să respecte o „abstinență absolută și continuă, lună confirmată Dacă nu a fost deja inițiată o metodă contraceptivă eficientă, pacientul trebuie îndrumat la un medic specialist pentru a stabili o metodă contraceptivă eficientă.
Mai jos sunt exemple de metode contraceptive considerate adecvate:
• Plantă
• Sistem intrauterin cu eliberare de levonorgestrel (IUS)
• Depozit de acetat de medroxiprogesteronă
• Sterilizarea tubară
• Raport sexual numai cu un partener masculin vasectomizat; vasectomia trebuie confirmată prin două analize negative ale materialului seminal
• Pastile numai cu progestin pentru inhibarea ovulației (de exemplu, desogestrel)
Datorită riscului crescut de tromboembolism venos la pacienții cu mielom multiplu care iau lenalidomidă în regimuri combinate și într-o măsură mai mică la pacienții cu sindroame mielodisplazice și limfom cu celule de manta care iau lenalidomidă în monoterapie, nu este recomandată utilizarea contraceptivelor orale. de asemenea secțiunea 4.5). Dacă pacientul ia în prezent un contraceptiv oral combinat, ar trebui să înlocuiască metoda controlului nașterii cu una dintre cele de mai sus. Riscul de tromboembolism venos rămâne timp de 4-6 săptămâni după întreruperea contraceptivului oral combinat. Eficacitatea steroizilor contraceptivi poate fi redusă în timpul tratamentului concomitent cu dexametazonă (vezi pct. 4.5).
Implanturile și sistemele intrauterine care eliberează levonorgestrel sunt asociate cu un risc crescut de infecție la inserare, precum și cu sângerări vaginale neregulate. Trebuie luată în considerare profilaxia antibiotică, în special la pacienții cu neutropenie.
Dispozitivele intrauterine care eliberează cupru nu sunt, în general, recomandate din cauza riscului potențial de infecție din momentul inserării și din cauza pierderii menstruale de sânge, care poate afecta negativ pacienții cu neutropenie sau trombocitopenie.
Test de sarcina
În conformitate cu practica locală, la pacienții cu vârsta fertilă, testele de sarcină cu o sensibilitate minimă de 25 mIU / ml trebuie efectuate sub supraveghere medicală, așa cum este descris mai jos. Această obligație este valabilă și pentru pacienții cu vârsta fertilă care practică abstinență absolută și continuă. În mod ideal, testul de sarcină, prescrierea și eliberarea medicamentului ar trebui să aibă loc în aceeași zi. Lenalidomida trebuie administrată pacienților cu vârsta fertilă în termen de 7 zile de la data prescripției.
Înainte de a începe tratamentul
După ce pacientul folosește contracepția eficientă timp de cel puțin 4 săptămâni, trebuie efectuat un test de sarcină supravegheat din punct de vedere medical în timpul consultației în care este prescris lenalidomida sau în cele 3 zile anterioare vizitei la medic. Testul trebuie să se asigure că pacienta nu este însărcinată înainte de a începe tratamentul cu lenalidomidă.
Urmărirea și sfârșitul tratamentului
Un test de sarcină supravegheat medical trebuie repetat la fiecare 4 săptămâni, inclusiv la 4 săptămâni după terminarea tratamentului, cu excepția cazurilor de sterilizare tubară confirmată. Aceste teste de sarcină trebuie efectuate în aceeași zi cu prescripția medicului sau în cele 3 zile anterioare vizitei medicului.
Pacienți bărbați
În timpul tratamentului, lenalidomida este prezentă la niveluri extrem de scăzute în material seminal și este nedetectabilă în materialul seminal al subiecților sănătoși la 3 zile după întreruperea tratamentului (vezi pct. 5.2). Ca măsură de precauție și luând în considerare populațiile speciale de pacienți cu un timp de eliminare prelungit, cum ar fi pacienții cu insuficiență renală, toți pacienții de sex masculin care iau lenalidomidă ar trebui să folosească prezervative pe toată durata tratamentului, în timp ce întrerup tratamentul. terapie, dacă partenerul dvs. este însărcinat sau poate avea fertilitate și nu folosește contracepție eficientă (chiar dacă bărbatul a suferit o vasectomie).
Precauții suplimentare pentru utilizare
Pacienții trebuie instruiți să nu dea niciodată acest medicament altor persoane și să returneze capsulelor neutilizate farmacistului la sfârșitul tratamentului.
Pacienții nu trebuie să doneze sânge în timpul tratamentului cu lenalidomidă și timp de cel puțin o săptămână după întreruperea tratamentului.
Materiale educaționale, restricții de prescripție și distribuire
Pentru a ajuta pacienții să evite expunerea fetală la lenalidomidă, deținătorul autorizației de punere pe piață va furniza materiale educaționale personalului medical pentru a consolida avertismentele privind teratogenitatea preconizată a lenalidomidei, pentru a oferi consiliere privind contracepția înainte de inițierea terapiei și pentru a oferi îndrumări cu privire la necesitatea unui test de sarcină. . Medicul trebuie să informeze pacienții bărbați și femeile despre riscul teratogen și măsurile stricte de prevenire a sarcinii, așa cum se specifică în Programul de prevenire a sarcinii, și să furnizeze pacienților broșura educațională adecvată, cardul pacientului și / sau instrumentul echivalent, în conformitate cu măsurile implementate la la nivel național. Un sistem național de control al distribuției a fost implementat în colaborare cu fiecare autoritate națională competentă. Acest sistem prevede utilizarea unui card de pacient și / sau a unui instrument echivalent, pentru controlul prescripției și eliberării, precum și colectarea datelor detaliate referitoare la indicație, pentru a controla cu atenție utilizarea fără etichetă medicamentului pe teritoriul național. În mod ideal, testul de sarcină, eliberarea rețetei și eliberarea medicamentului ar trebui să aibă loc în aceeași zi. Lenalidomida trebuie administrată pacienților cu vârsta fertilă în termen de 7 zile de la data prescripției. iar după „rezultatul negativ al testului de sarcină efectuat sub supraveghere medicală.
Avertismente speciale suplimentare și precauții de utilizare
Tulburări cardiovasculare
Infarct miocardic
Au fost observate cazuri de infarct miocardic la pacienții cărora li s-a administrat lenalidomidă, în special la cei cu factori de risc cunoscuți, și în primele 12 luni, administrate în asociere cu dexametazonă. Pacienții cu factori de risc cunoscuți, inclusiv cei cu tromboză anterioară, trebuie monitorizați îndeaproape și trebuie luate măsuri pentru a încerca să minimizeze toți factorii de risc modificabili (de exemplu fumatul, hipertensiunea și hiperlipidemia).
Evenimente tromboembolice venoase și arteriale
La pacienții cu mielom multiplu, combinația de lenalidomidă și dexametazonă este asociată cu un risc crescut de tromboembolism venos (în principal tromboză venoasă profundă și embolie pulmonară) și tromboembolism arterial (în principal infarct miocardic și eveniment cerebrovascular). extinderea cu lenalidomidă în asociere cu melfalan și prednison în mielomul multiplu nou diagnosticat și ca monoterapie în sindroamele mielodisplazice Vezi secțiunile 4.5 și 4.8.
La pacienții cu sindroame mielodisplazice și limfom cu celule de manta, tratamentul numai cu lenalidomidă a fost asociat și cu un risc de tromboembolism venos (în principal tromboză venoasă profundă și embolie pulmonară), dar într-o măsură mai mică decât la pacienții cu mielom multiplu - vezi pct. 4.5 și 4.8.
Prin urmare, pacienții cu factori de risc cunoscuți pentru tromboembolism - inclusiv o tromboză anterioară - trebuie monitorizați îndeaproape. Trebuie luate măsuri pentru a încerca să minimizeze toți factorii de risc modificabili (de exemplu fumatul, hipertensiunea și hiperlipidemia). La acești pacienți, administrarea concomitentă de agenți eritropoietici sau antecedente de evenimente tromboembolice poate crește, de asemenea, riscul de tromboză. Prin urmare, la pacienții cu mielom multiplu care iau lenalidomidă și dexametazonă, se recomandă ca agenți eritropoietici sau alți agenți care pot crește riscul de tromboză, cum ar fi de exemplu. terapie de înlocuire a hormonilor. Dacă concentrația de hemoglobină crește peste 12 g / dl, utilizarea agenților eritropoietici trebuie întreruptă.
Pacienții și medicii trebuie să fie conștienți de necesitatea de a acorda atenție semnelor și simptomelor tromboembolismului. Pacienții trebuie să solicite asistență medicală dacă apar simptome precum scurtarea respirației, dureri în piept, umflarea membrelor inferioare sau superioare. În scopuri profilactice, trebuie recomandat aportul de medicamente antitrombotice, în special la pacienții cu factori de risc trombotici suplimentari. Decizia de a adopta măsuri antitrombotice profilactice trebuie luată după o analiză atentă a factorilor de risc pentru fiecare pacient în parte.
Dacă pacientul are vreun eveniment tromboembolic, tratamentul trebuie întrerupt și trebuie inițiat tratamentul anticoagulant standard. Odată ce pacientul a fost stabilizat după anticoagulare și toate complicațiile evenimentului tromboembolic s-au rezolvat, tratamentul cu lenalidomidă poate fi reluat la doza inițială după o evaluare a raportului beneficiu-risc. Pacientul trebuie să continue terapia anticoagulantă în timpul tratamentului.
Neutropenie și trombocitopenie
Principalele toxicități ale lenalidomidei care limitează doza includ neutropenia și trombocitopenia. Pentru a monitoriza posibila apariție a citopeniei, ar trebui efectuată o numărare completă de globule albe, inclusiv număr de globule albe, inclusiv diferențial, număr de trombocite, hemoglobină și hematocrit, la momentul inițial, o dată pe săptămână, în primele 8 săptămâni de tratament. La pacienții cu limfom cu celule de manta, programul de monitorizare trebuie să fie la fiecare 2 săptămâni în ciclurile 3 și 4 și la începutul fiecărui ciclu ulterior. Poate fi necesară o reducere a dozei (vezi pct. 4.2). În caz de neutropenie, medicul ar trebui să ia în considerare utilizarea factorilor de creștere în gestionarea pacientului. Pacienții trebuie sfătuiți să raporteze imediat episoadele febrile. Se recomandă prudență în administrarea concomitentă a lenalidomidei cu alți agenți mielosupresori.
• Mielom multiplu nou diagnosticat la pacienții tratați cu lenalidomidă în asociere cu doză mică de dexametazonă
Neutropenia de gradul 4 a fost observată într-o măsură mai mică în lenalidomidă în combinație cu brațele de tratament cu doxametazonă în doză mică comparativ cu brațul comparator (8,5% în Rd [tratament continuu] și Rd18 [tratament pentru 18 cicluri de patru săptămâni], comparativ cu 15 % în brațul melfalan / prednison / talidomidă, vezi pct. 4.8). Episoadele de neutropenie febrilă de gradul 4 au fost în concordanță cu brațul comparativ (0,6% la pacienții tratați cu lenalidomidă / dexametazonă Rd și Rd18, comparativ cu 0,7% la pacienții din brațul melfalan / prednison / talidomidă, vezi pct. 4.8). Pacienții trebuie sfătuiți să raporteze imediat episoadele febrile și poate fi necesară reducerea dozei (vezi pct. 4.2).
Trombocitopenia de gradul 3 sau 4 a fost observată într-o măsură mai mică în brațele Rd și Rd18 decât în brațul comparator (8,1% față de 11,1%, respectiv). Pacienții și medicii trebuie să respecte semnele și simptomele sângerării, inclusiv peteșii și epistaxis, în special la pacienții supuși unui tratament concomitent care ar putea induce sângerări (vezi pct. 4.8, Tulburări de sângerare).
• Mielom multiplu nou diagnosticat la pacienții tratați cu lenalidomidă în asociere cu melfalan și prednison
În studiile clinice la pacienți cu mielom multiplu nou diagnosticat, combinația de lenalidomidă cu melfalan și prednison este asociată cu o incidență mai mare a neutropeniei de gradul 4 (34,1% la pacienții din brațul melfalan, prednison și lenalidomidă urmată de lenalidomidă [MPR + R]) și melfalan, prednison și lenalidomidă urmate de placebo [MPR + p], comparativ cu 7,8% dintre pacienții tratați cu MPp + p; vezi pct. 4.8). R / MPR + p, comparativ cu 0,0% la pacienții tratați cu MPp + p; vezi pct. 4.8).
La pacienții cu mielom multiplu, combinația de lenalidomidă cu melfalan și prednison este asociată cu o incidență mai mare a trombocitopeniei de gradul 3 și gradul 4 (40,4% la pacienții tratați cu MMR + R / MMR + p, comparativ cu 13,7% la pacienții tratați cu MPp + p; vezi pct. 4.8) Pacienții și medicii trebuie să fie atenți la semne și simptome de sângerare, inclusiv petechii și epistaxis, în special la pacienții tratați concomitent cu medicamente care cresc predispoziția la sângerare (vezi pct. 4.8, Tulburări de sângerare).
• Mielom multiplu cu cel puțin o terapie anterioară
La pacienții cu mielom multiplu care primesc cel puțin un tratament anterior, combinația de lenalidomidă și dexametazonă este asociată cu o incidență mai mare a neutropeniei de gradul 4 (5,1% dintre pacienții tratați cu lenalidomidă / dexametazonă comparativ cu 0,6% dintre pacienții tratați cu placebo / dexametazonă; vezi pct. 4.8). Episoadele de neutropenie febrilă de gradul 4 au fost observate rar (la 0,6% dintre pacienții tratați cu lenalidomidă / dexametazonă comparativ cu 0,0% dintre pacienții tratați cu placebo / dexametazonă; vezi pct. 4.8) Pacienții trebuie sfătuiți să raporteze imediat episoade febrile Poate fi necesară reducerea dozelor (vezi pct. 4.2). În caz de neutropenie, medicii ar trebui să ia în considerare utilizarea factorilor de creștere în gestionarea pacientului.
La pacienții cu mielom multiplu, combinația de lenalidomidă și dexametazonă este asociată cu o incidență mai mare a trombocitopeniei de gradul 3 și a gradului 4 (9,9% și respectiv 1,4%, incidența mai mare a trombocitopeniei de gradul 3) .3 și Gradul 4 (9,9%) și 1,4% dintre pacienții tratați cu lenalidomidă / respectiv dexametazonă, comparativ cu 2,3% și 0,0% dintre pacienții tratați cu placebo / dexametazonă; tratate concomitent cu medicamente care pot induce sângerări (vezi pct. 4.8, Tulburări de sângerare).
• Sindroame mielodisplazice
La pacienții cu sindroame mielodisplazice, tratamentul cu lenalidomidă este asociat cu o incidență mai mare de neutropenie și trombocitopenie de gradul 3 și 4 decât la pacienții tratați cu placebo (vezi pct. 4.8).
• Limfom cu celule de manta
La pacienții cu limfom cu celule de manta, tratamentul cu lenalidomidă este asociat cu o incidență mai mare a neutropeniei de gradul 3 și 4 decât la pacienții din brațul de control (vezi pct. 4.8).
Infecție cu sau fără neutropenie
Pacienții cu mielom multiplu sunt predispuși la apariția infecțiilor, inclusiv a pneumoniei. O rată mai mare de infecții a fost observată în timpul tratamentului cu lenalidomidă în asociere cu dexametazonă decât cu MPT. Infecții de grad ≥ 3 au apărut în contextul neutropeniei la mai puțin de o treime din pacienți. Pacienții cu factori de risc cunoscuți pentru infecții trebuie monitorizați îndeaproape. Toți pacienții trebuie sfătuiți să consulte medicul imediat la primul semn de infecție (de exemplu, tuse, febră etc.), pentru a permite o intervenție promptă pentru a reduce severitatea.
Insuficiență renală
Lenalidomida este substanțial excretată prin rinichi. Prin urmare, la pacienții cu insuficiență renală trebuie acordată o atenție deosebită în alegerea dozei și este recomandabilă monitorizarea funcției renale (vezi pct. 4.2).
Tulburări ale glandei tiroide
Au fost observate cazuri de hipotiroidism și hipertiroidism. Înainte de începerea tratamentului, se recomandă controlul optim al comorbidităților care afectează funcția tiroidiană. Se recomandă monitorizarea funcției tiroidiene la momentul inițial și în timpul tratamentului.
Neuropatie periferica
Lenalidomida este legată structural de talidomidă, despre care se știe că provoacă neuropatie periferică severă. Nu s-a observat o creștere a neuropatiei periferice cu utilizarea pe termen lung a lenalidomidei pentru tratamentul mielomului multiplu nou diagnosticat.
Reacția de erupție tumorală și sindromul de liză tumorală
Deoarece lenalidomida prezintă activitate anti-neoplazică, complicațiile sindromului de liză tumorală (Sindromul de liză tumorală, TLS). TLS și Reacție de erupție tumorală (TFR) au fost frecvent observate la pacienții cu leucemie limfocitară cronică (LLC) și observate neobișnuit la pacienții cu limfoame tratați cu lenalidomidă. În timpul tratamentului cu lenalidomidă au fost raportate cazuri de TLS cu rezultat fatal. Pacienții cu risc de TLS și TFR sunt cei cu sarcină tumorală mare înainte de tratament. La acești pacienți, trebuie inițiată precauție la inițierea tratamentului cu lenalidomidă. Se recomandă ca acești pacienți să fie monitorizați cu atenție, în special în timpul primului ciclu sau creșterea dozei, și să se ia măsurile de precauție adecvate. Au fost raportate cazuri rare de TLS la pacienții cu MM tratați cu lenalidomidă, în timp ce nu au fost raportate cazuri la pacienții cu MDS tratat cu lenalidomidă.
Masa tumorală
• Limfom cu celule de manta
Lenalidomida nu este recomandată pentru tratamentul pacienților cu sarcină tumorală mare dacă sunt disponibile opțiuni de tratament alternative.
Moarte prematura
În studiul MCL-002 a existat o creștere evidentă generală a deceselor timpurii (în decurs de 20 de săptămâni). Pacienții cu sarcină tumorală inițială ridicată au un risc mai mare de deces precoce: au existat 16/81 (20%) decese precoce în brațul cu lenalidomidă și 2/28 (7%) decese precoce în brațul de control. Până la 52 de săptămâni, cifrele corespunzătoare erau 32/81 (40%) și 6/28 (21%) (vezi secțiunea 5.1).
Evenimente adverse
În studiul MCL-002 în timpul ciclului 1 de tratament, 11/81 (14%) pacienți cu sarcină tumorală mare au fost retrași din terapia cu lenalidomidă, comparativ cu 1/28 (4%) în grupul de control. Motivul principal pentru întreruperea tratamentului pentru pacienții cu sarcină tumorală ridicată în timpul ciclului 1 de tratament în brațul cu lenalidomidă s-a datorat evenimentelor adverse, 7/11 (64%).
De aceea, pacienții cu sarcină tumorală ridicată trebuie monitorizați cu atenție pentru reacții adverse (vezi pct. 4.8), inclusiv orice semne de Reacție de erupție tumorală (TFR). Pentru ajustări ale dozei în cazul TFR, vezi pct. 4.2.
O masă tumorală crescută a fost definită ca cel puțin o leziune ≥ 5 cm în diametru sau 3 leziuni ≥ 3 cm.
Reacție de erupție tumorală
• Limfom cu celule de manta
Se recomandă monitorizarea și evaluarea atentă a TFR. Pacienții cu MIPI crescut (Mantel Cell Lymphoma International Prognostic Index) la diagnostic sau boală caracterizată prin mase tumorale mari (cel puțin o leziune care este ≥ 7 cm în cel mai lung diametru) la momentul inițial poate prezenta un risc pentru TFR. Acolo Reacție de erupție tumorală poate simula progresia bolii (PD). Pacienții din studiile MCL-002 și MCL-001 care au prezentat TFR de gradul 1 și 2 au fost tratați cu corticosteroizi, antiinflamatoare nesteroidiene (AINS) și / sau analgezice narcotice pentru gestionarea simptomelor TFR. Decizia de a adopta măsuri terapeutice pentru TFR trebuie luată după o „evaluare clinică atentă a pacientului individual (vezi pct. 4.2).
Reactii alergice
Au fost raportate cazuri de reacții alergice / de hipersensibilitate la pacienții tratați cu lenalidomidă (vezi pct. 4.8). Se recomandă monitorizarea atentă a pacienților care au avut reacții alergice anterioare la talidomidă, deoarece în literatura de specialitate a fost raportată o posibilă reacție încrucișată între lenalidomidă și talidomidă.
Reacții cutanate severe
Au fost raportate cazuri de SSJ și NET. Tratamentul cu lenalidomidă trebuie întrerupt în cazul unei erupții exfoliative sau buloase sau în cazul în care se suspectează SSJ sau NET și nu trebuie reluat după întreruperea acesteia din cauza acestor reacții. Întreruperea sau întreruperea tratamentului cu lenalidomidă trebuie luată în considerare pentru alte forme de reacții cutanate, în funcție de severitatea acestora.Pacienții cu antecedente de erupții cutanate severe asociate tratamentului cu talidomidă nu trebuie să primească lenalidomidă.
Intoleranță la lactoză
Capsulele Revlimid conțin lactoză. Pacienții cu probleme ereditare rare de intoleranță la galactoză, deficit de lactază Lapp sau malabsorbție la glucoză-galactoză nu trebuie să ia acest medicament.
Capsule neutilizate
Pacienții trebuie sfătuiți să nu dea niciodată acest medicament altor persoane și să returneze capsulelor neutilizate farmacistului la sfârșitul tratamentului.
A doua tumori primare
O creștere a tumorilor primare secundare (A doua primară Malignitate, SPM) la pacienții cu mielom tratați anterior cu lenalidomidă / dexametazonă (3,98 la 100 persoane-ani) versus martori (1,38 la 100 persoane-ani). SPM neinvazive constau în carcinoame cu celule bazale sau cu celule scuamoase.
Cele mai multe SPM invazive erau tumori solide.
În studiile clinice efectuate la pacienții nou diagnosticați cu mielom multiplu neeligibili pentru transplant, a fost observată o creștere de 4,9 ori a ratei incidenței PMS hematologic (cazuri de LMA, SMD) la pacienții tratați cu lenalidomidă în asociere cu melfalan și prednison până la progresie (1,75 pe 100 persoane-ani), comparativ cu melfalan în combinație cu prednison (0,36 la 100 pe persoană-ani).
O creștere de 2,12 ori a ratei de incidență a SPM solid a fost observată la pacienții tratați cu lenalidomidă (9 cicluri) în asociere cu melfalan și prednison (1,57 la 100 persoane-ani), comparativ cu melfalan în combinație cu prednison (0,74 la 100 pe persoană-ani).
La pacienții tratați cu lenalidomidă în asociere cu dexametazonă până la progresie sau timp de 18 luni, rata incidenței sindromului premenstrual hematologic (0,16 la 100 de ani-persoană) nu a fost crescută în comparație cu talidomida în combinație cu melfalan și prednison (0,79 la 100 de ani-persoană) .
O creștere de 1,3 ori a ratei de incidență a sindromului premenstrual solid a fost observată la pacienții tratați cu lenalidomidă în asociere cu dexametazonă până la progresie sau timp de 18 luni (1,58 la 100 persoane-ani) comparativ cu talidomida în combinație cu melfalan și prednison (1,19 la 100 persoană-ani).
În studiile clinice la pacienții nou diagnosticați cu mielom multiplu eligibili pentru transplant, a fost observată o rată crescută a incidenței sindromului premenstrual hematologic la pacienții tratați cu lenalidomidă imediat după doze mari de melfalan și transplant autolog de celule stem (Transplant de celule stem autologe, ASCT), comparativ cu pacienții tratați cu placebo (1,27-1,56 și 0,46-0,53 la 100 persoane-ani, respectiv). Cazurile de tumori maligne cu celule B (inclusiv limfomul Hodgkin) observate în studiile clinice au fost la pacienții tratați cu lenalidomidă în post-ASCT.
Riscul apariției sindromului premenstrual hematologic trebuie luat în considerare înainte de inițierea tratamentului cu Revlimid în asociere cu melfalan sau în perioada imediat următoare dozei mari de melfalan și ASCT. Medicii trebuie să evalueze cu atenție pacienții înainte și în timpul tratamentului, utilizând screening-ul standard al cancerului pentru sindromul premenstrual și să instituie tratamentul conform instrucțiunilor.
Progresia către leucemie mieloidă acută (LMA) în sindromul mielodisplazic (SMD) la risc scăzut sau intermediar-1
• Cariotip
Variabilele de bază, inclusiv anomalii citogenetice complexe, sunt asociate cu progresia la LMA la subiecții dependenți de transfuzie cu anomalie de deleție 5q izolată. Într-o analiză combinată a două studii clinice efectuate cu Revlimid în SMD cu risc scăzut sau intermediar 1, subiecții cu anomalii citogenetice complexe au avut cel mai mare risc cumulativ de progresie la LMA estimat la 2 ani (38,6%). Rata estimată de progresie pe 2 ani la LMA la pacienții cu anomalie de deleție 5q izolată a fost de 13,8%, comparativ cu 17,3% la pacienții cu anomalie de deleție 5q izolată și o „anomalie citogenetică suplimentară.
În consecință, raportul beneficiu / risc al Revlimid este necunoscut atunci când MDS este asociat cu anomalii de deleție 5q izolate și anomalii citogenetice complexe.
• Starea TP53
O mutație TP53 este prezentă la 20-25% dintre pacienții cu SMD cu anomalie de deleție izolată 5q cu risc scăzut și este asociată cu un risc mai mare de progresie la LMA. Într-o „analiză post-hoc a unui studiu clinic (MDS-004) efectuat cu Revlimid în SMD cu risc scăzut sau intermediar 1, rata estimată de progresie pe 2 ani către LMA a fost de 27,5% la pacienții cu IHC-p53 pozitiv (1 % tăiere a colorării nucleare puternice, utilizând evaluarea imunohistochimică a proteinei p53 ca surogat pentru starea mutației TP53) și 3,6% la pacienții cu IHC-p53 negativ (p = 0,0038) (vezi secțiunea 4.8).
Progresia către alte tumori maligne în limfomul cu celule de manta
În limfomul cu celule de manta, LMA, tumorile maligne cu celule B și cancerul de piele non-melanom (NMSC) sunt riscuri potențiale.
Tulburări hepatice
Au fost observate cazuri de insuficiență hepatică, inclusiv cu rezultat fatal, la pacienții tratați cu lenalidomidă în terapia combinată: insuficiență hepatică acută, hepatită toxică, hepatită citolitică, hepatită colestatică și hepatită citolitică / colestatică mixtă. Mecanismele hepatotoxicității severe induse de medicamente rămân necunoscute, deși, în unele cazuri, factorii de risc pot fi boli hepatice virale preexistente, enzime hepatice de bază crescute și, eventual, tratament cu antibiotice.
Au fost observate în mod obișnuit anomalii ale testelor funcției hepatice și au fost în general asimptomatice și reversibile la întreruperea tratamentului. Odată ce parametrii au revenit la valorile inițiale, poate fi luată în considerare reluarea tratamentului la o doză mai mică.
Lenalidomida se excretă prin rinichi. Este important să se ajusteze doza la pacienții cu insuficiență renală pentru a evita atingerea concentrațiilor plasmatice care ar putea crește riscul unor reacții adverse hematologice mai importante sau hepatotoxicitate. Se recomandă monitorizarea funcției hepatice, în special în cazul unei infecții hepatice virale anterioare sau concomitente sau când lenalidomida este administrată în asociere cu medicamente cunoscute a fi asociate cu disfuncție hepatică.
Pacienți cu mielom multiplu nou diagnosticat
A existat o rată mai mare de intoleranță (evenimente adverse de gradul 3 sau 4, evenimente adverse grave, întreruperea tratamentului) la pacienții cu vârsta> 75 de ani, stadiu ISS (Sistem internațional de etapizare) III, starea de performanță (PS) ≤ 2 evaluată conform criteriilor ECOG (Grupul de Oncologie Cooperativă din Est) sau CLcr
Cataractă
Cataracta a fost observată mai frecvent la pacienții tratați cu lenalidomidă în asociere cu dexametazonă, în special atunci când este utilizată pentru o perioadă prelungită. Se recomandă monitorizarea periodică a capacității vizuale.
04.5 Interacțiuni cu alte medicamente și alte forme de interacțiune -
Agenții eritropoietici sau alți agenți care pot crește riscul de tromboză, cum ar fi terapia de substituție hormonală, trebuie utilizați cu precauție la pacienții cu mielom multiplu care iau lenalidomidă și dexametazonă (vezi pct. 4.4 și 4.8). administrarea de lenalidomidă și dexametazonă (vezi pct. 4.4 și 4.8).
Contraceptive orale
Nu s-au efectuat studii de interacțiune cu contraceptive orale. Lenalidomida nu este un inductor enzimatic. Într-un studio in vitro efectuate cu hepatocite umane, lenalidomida, testată la diferite concentrații, nu a indus CYP1A2, CYP2B6, CYP2C9, CYP2C19 și CYP3A4 / 5. Prin urmare, dacă lenalidomida se administrează singură, nu este de așteptat inducția care duce la reducerea eficacității medicamentelor, inclusiv a contraceptivelor hormonale. Cu toate acestea, se știe că dexametazona este un inductor slab până la moderat al CYP3A4 și care poate afecta alte enzime și proteine de transport. Nu este exclus ca eficacitatea contraceptivelor orale să fie redusă în timpul tratamentului.
Trebuie luate măsuri eficiente pentru a evita sarcina (vezi pct. 4.4 și 4.6).
Warfarina
Administrarea concomitentă de 10 mg doze repetate de lenalidomidă nu a avut niciun efect asupra farmacocineticii unei doze unice de R- și S-warfarină. Administrarea concomitentă a unei singure doze de warfarină de 25 mg nu a avut niciun efect asupra farmacocineticii lenalidomidei. Cu toate acestea, nu se știe dacă există o „interacțiune în timpul” utilizării clinice (tratament concomitent cu dexametazona). Dexametazona este un inductor enzimatic slab până la moderat, iar efectele sale asupra warfarinei sunt necunoscute. Se recomandă monitorizarea atentă a concentrației de warfarină în timpul tratamentului.
Digoxină
Administrarea concomitentă de 10 mg / zi de lenalidomidă a crescut concentrația plasmatică a digoxinei cu 14% (0,5 mg, doză unică) cu un CI (interval de încredere) de 90% [0,52% -28,2%]. Nu se știe dacă efectul ar diferi în situația terapeutică (doze mai mari de lenalidomidă și tratament concomitent cu dexametazonă). Prin urmare, se recomandă monitorizarea concentrației digoxinei în timpul tratamentului cu lenalidomidă.
Statine
Când statinele sunt administrate cu lenalidomidă, există un risc crescut de rabdomioliză, care poate fi pur și simplu aditivă. Este necesară o monitorizare clinică și de laborator îmbunătățită, în special în primele săptămâni de tratament.
Dexametazona
Administrarea concomitentă de doze unice sau multiple de dexametazonă (40 mg / zi) nu are efect clinic relevant asupra farmacocineticii cu doze multiple de lenalidomidă (25 mg / zi).
Interacțiuni cu inhibitori ai glicoproteinei P (P-gp)
In vitro, Lenalidomida este un substrat al P-gp, dar nu este un inhibitor al P-gp. Administrarea concomitentă a mai multor doze de inhibitor puternic al P-gp, chinidină (600 mg, de două ori pe zi) sau inhibitor al P-gp / substrat temsirolimus cu acțiune moderată (25 mg), nu are efect clinic relevant asupra farmacocineticii lenalidomidei (25 mg) Administrarea concomitentă de lenalidomidă nu modifică farmacocinetica temsirolimus.
04.6 Sarcina și alăptarea -
Femeile aflate la vârsta fertilă / Contracepția la bărbați și femei
Femeile aflate la vârsta fertilă trebuie să utilizeze metode contraceptive eficiente. Dacă sarcina apare în timpul tratamentului cu lenalidomidă, tratamentul trebuie întrerupt și pacientul trebuie să meargă la un specialist sau cu experiență în teratologie, care poate evalua situația și poate da o opinie. Dacă partenerul unui pacient de sex masculin care ia lenalidomidă este gravidă, partenerul trebuie sfătuit să meargă la un medic specialist sau medic cu experiență în teratologie, care poate evalua situația și poate da o opinie.
În timpul tratamentului, lenalidomida este prezentă la niveluri extrem de scăzute în material seminal și este nedetectabilă în materialul seminal al subiecților sănătoși la 3 zile după întreruperea tratamentului (vezi pct. 5.2). Ca măsură de precauție și luând în considerare populațiile speciale de pacienți cu un timp de eliminare prelungit, cum ar fi pacienții cu insuficiență renală, toți pacienții de sex masculin care iau lenalidomidă trebuie să utilizeze prezervative pe toată durata tratamentului, în timpul suspendării dozei și până la o săptămână după întreruperea tratamentului dacă partenerul dvs. este gravidă sau poate avea fertilitate și nu utilizează metode contraceptive.
Sarcina
Lenalidomida este legată structural de talidomidă, o substanță activă cu un efect teratogen cunoscut la om, care provoacă malformații congenitale grave care pun viața în pericol.
Lenalidomida a indus malformații la maimuțe similare cu cele descrise pentru talidomidă (vezi pct. 5.3). Prin urmare, este de așteptat un efect teratogen al lenalidomidei, iar lenalidomida este contraindicată în timpul sarcinii (vezi pct. 4.3).
Timp de hrănire
Deoarece nu se știe dacă lenalidomida este excretată în laptele matern uman, se recomandă întreruperea alăptării în timpul tratamentului cu lenalidomidă.
Fertilitate
Un studiu de fertilitate efectuat la șobolani cu doze de lenalidomidă de până la 500 mg / kg (aproximativ de 200 până la 500 de ori mai mare decât dozele de 25 mg și respectiv 10 mg, utilizate la om și calculate pe baza suprafeței corporale), nu a arătat efecte adverse asupra fertilitate sau toxicitate maternă.
04.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje -
Lenalidomida are un efect ușor sau moderat asupra capacității de a conduce vehicule și de a folosi utilaje. În timpul tratamentului cu lenalidomidă au fost raportate oboseală, amețeli, somnolență, amețeli și vedere încețoșată. Prin urmare, se recomandă precauție atunci când conduceți vehicule sau folosiți utilaje.
04.8 Efecte nedorite -
Rezumatul profilului de siguranță
Mielom multiplu nou diagnosticat la pacienții tratați cu lenalidomidă în asociere cu doze mici de dexametazona
Cele mai frecvent observate (≥ 5%) reacții adverse grave cu lenalidomidă în asociere cu doze mici de dexametazonă (Rd și Rd18), comparativ cu melfalan, prednison și talidomidă (MPT), au fost:
• Pneumonie (9,8%)
• Insuficiență renală (inclusiv acută) (6,3%)
Reacțiile adverse observate mai frecvent cu Rd sau Rd18 decât cu MPT au fost: diaree (45,5%), oboseală (32,8%), dureri de spate (32,0%), astenie (28,2%), insomnie (27,6%), erupții cutanate (24,3%) , scăderea poftei de mâncare (23,1%), tuse (22,7%), pirexie (21,4%) și spasme musculare (20,5%).
Pacienți nou diagnosticați cu mielom multiplu tratați cu lenalidomidă în asociere cu melfalan și prednison
Cele mai frecvent observate reacții adverse grave (≥ 5%) cu melfalan, prednison și lenalidomidă urmate de terapia de întreținere cu lenalidomidă (MPR + R) sau melfalan, prednison și lenalidomidă urmată de placebo (MPR + p), comparativ cu melfalan, prednison și placebo urmat de placebo (MPp + p), au fost:
• Neutropenie febrilă (6,0%)
• Anemie (5,3%)
Reacțiile adverse observate mai frecvent cu MPR + R sau MPR + p decât cu MPp + p au fost: neutropenie (83,3%), anemie (70,7%), trombocitopenie (70,0%), leucopenie (38, 8%), constipație (34,0 %), diaree (33,3%), erupție cutanată (28,9%), pierexie (27,0%), edem periferic (25,0%), tuse (24,0%), apetit scăzut (23,7%) și astenie (22,0%).
Mielom multiplu cu cel puțin o terapie anterioară
În două studii de fază III controlate cu placebo, 353 de pacienți cu mielom multiplu au fost expuși tratamentului combinat cu lenalidomidă / dexametazonă și 351 la tratament combinat cu placebo / dexametazonă.
Cele mai grave reacții adverse observate mai frecvent la combinația lenalidomidă / dexametazona decât la combinația placebo / dexametazonă au fost:
• Tromboembolism venos (tromboză venoasă profundă, embolie pulmonară) (vezi pct. 4.4)
• Neutropenie de gradul 4 (vezi pct. 4.4).
Reacțiile adverse care au apărut mai frecvent cu lenalidomidă și dexametazonă, comparativ cu placebo și dexametazonă, la combinarea studiilor clinice cu mielom multiplu (MM-009 și MM-010), au fost oboseală (43,9%), neutropenie (42,2%), constipație ( 40,5%), diaree (38,5%), crampe musculare (33,4%), anemie (31,4%), trombocitopenie (21,5%) și erupții cutanate (21, 2%).
Sindroame mielodisplazice
Profilul general de siguranță al Revlimid la pacienții cu sindroame mielodisplazice se bazează pe date de la un total de 286 de pacienți incluși într-un studiu de fază II și de fază III (vezi pct. 5.1). În faza II, toți cei 148 de pacienți au fost tratați cu lenalidomidă. În studiul de fază III, 69 de pacienți au fost tratați cu lenalidomidă 5 mg, 69 pacienți au fost tratați cu lenalidomidă 10 mg și 67 de pacienți au primit placebo în timpul fazei dublu-orb a studiului.
Majoritatea reacțiilor adverse au avut tendința să apară în primele 16 săptămâni de tratament cu lenalidomidă.
Reacțiile adverse grave includ:
• Tromboembolism venos (tromboză venoasă profundă, embolie pulmonară) (vezi pct. 4.4)
• Neutropenie de gradul 3 sau 4, neutropenie febrilă și trombocitopenie de gradul 3 sau 4 (vezi pct. 4.4).
Cele mai frecvent observate reacții adverse care au apărut mai frecvent în grupurile cu lenalidomidă decât în brațul control (placebo) în studiul de fază III au fost neutropenia, (76,8%), trombocitopenia (46,4%)., Diareea (34,8%), constipația ( 19,6%), greață (19,6%), mâncărime (25,4%), erupție cutanată (18,1%), oboseală (18,1%) și spasme musculare (16,7%).
Limfom cu celule de manta
Profilul general de siguranță al Revlimid la pacienții cu limfom cu celule de manta se bazează pe datele de la 254 de pacienți incluși într-un studiu randomizat, controlat de fază II, MCL-002 (vezi pct. 5.1).
În plus, reacțiile adverse la medicamente (ADR) observate în studiul de susținere MCL-001 au fost incluse în Tabelul 3.
Cele mai frecvent observate reacții adverse grave în studiul MCL-002 (cu o diferență de cel puțin 2 puncte procentuale) la brațul cu lenalidomidă față de brațul de control au fost:
• Neutropenie (3,6%)
• Embolie pulmonară (3,6%)
• Diaree (3,6%)
Cele mai frecvent observate reacții adverse care au apărut mai frecvent în brațul de lenalidomidă decât în brațul de control din studiul MCL-002 au fost neutropenia, (50,9%), anemia (28,7%), diareea (22, 8%), oboseala (21,0%) , constipație (17,4%), pirexie (16,8%) și erupții cutanate (inclusiv dermatită alergică) (16,2%).
În studiul MCL-002 a existat o creștere evidentă generală a deceselor timpurii (în decurs de 20 de săptămâni). Pacienții cu sarcină tumorală inițială ridicată au un risc mai mare de deces precoce: 16/81 (20%) decese precoce în brațul cu lenalidomidă și 2/28 (7%) decese precoce în brațul de control. Până la 52 de săptămâni, cifrele corespunzătoare erau 32/81 (39,5%) și 6/28 (21%) (vezi secțiunea 5.1).
În timpul ciclului 1 de tratament, 11/81 (14%) pacienți cu sarcină tumorală mare au fost retrași din terapia cu lenalidomidă, comparativ cu 1/28 (4%) în grupul de control. Motivul principal pentru întreruperea tratamentului pentru pacienții cu sarcină tumorală ridicată în timpul ciclului 1 de tratament în brațul cu lenalidomidă s-a datorat evenimentelor adverse, 7/11 (64%).
O masă tumorală crescută a fost definită ca cel puțin o leziune ≥ 5 cm în diametru sau 3 leziuni ≥ 3 cm.
Lista sumară a reacțiilor adverse
Tabel rezumativ pentru terapia combinată
Reacțiile adverse observate la pacienții tratați pentru mielom multiplu sunt enumerate mai jos în funcție de clasa de sisteme de organe și de frecvență. În cadrul fiecărei clase de frecvență, reacțiile adverse sunt enumerate în ordinea descrescătoare a severității. Frecvențele sunt definite după cum urmează: foarte frecvente (≥ 1/10); frecvente (≥ 1/100,
Tabelul următor a fost compilat pe baza datelor colectate în timpul studiilor de mielom multiplu cu terapie combinată. Datele nu au fost actualizate luând în considerare durata mai lungă a tratamentului la brațele care conțin lenalidomidă a continuat până la progresia bolii, comparativ cu brațele comparative din studiile pivot de mielom multiplu (vezi pct. 5.1).
Reacțiile adverse au fost plasate în categoria corespunzătoare din tabelul de mai jos, în funcție de cea mai mare incidență observată în oricare dintre studiile clinice pivot.
Tabelul 1: Reacții adverse raportate în studiile clinice la pacienții cu mielom multiplu tratați cu lenalidomidă în asociere cu dexametazonă sau cu melfalan și prednison
^ Vezi secțiunea 4.8 Descrierea reacțiilor adverse selectate
* Cancerul de piele cu celule scuamoase a fost observat în studiile clinice la pacienții cu mielom tratați anterior cu lenalidomidă / dexametazonă comparativ cu martorii
** Cancerul de piele cu celule scuamoase a fost observat într-un studiu clinic la pacienții nou diagnosticați cu mielom cu lenalidomidă / dexametazonă comparativ cu martorii
Tabel rezumativ pentru monoterapie
Reacțiile adverse observate la pacienții tratați pentru sindroame mielodisplazice și limfom cu celule de manta sunt enumerate mai jos după clasa de sisteme de organe și frecvență.
În cadrul fiecărei clase de frecvență, reacțiile adverse sunt enumerate în ordinea descrescătoare a severității. Frecvența este definită ca: foarte frecvente (≥ 1/10); frecvente (≥ 1/100,
Următoarele tabele au fost compilate pe baza datelor colectate în timpul principalelor studii de monoterapie pentru sindroame mielodisplazice și limfom cu celule de manta.
Reacțiile adverse au fost plasate în categoria corespunzătoare în tabelele următoare, în funcție de cea mai mare incidență observată în oricare dintre studiile clinice pivot.
Tabelul 2: Reacții adverse raportate în studiile clinice la pacienții cu sindroame mielodisplazice tratați cu lenalidomidă
^ Vezi secțiunea 4.8 Descrierea reacțiilor adverse selectate
♦ Evenimente adverse observate ca fiind grave în studiile clinice cu sindroame mielodisplazice.
≈ Modificarea dispoziției a fost observată ca un eveniment advers comun frecvent în studiul sindroamelor mielodisplazice de fază III; nu a fost raportat ca un eveniment advers de gradul 3 sau 4.
Algoritm aplicat pentru intrarea SmPC: Toate reacțiile adverse la medicamente (ADR) capturate de algoritmul de studiu de fază III sunt incluse în SmPC european. Pentru aceste ADR-uri, a fost efectuat un control suplimentar al frecvenței ADR-urilor dobândite de algoritmul de studiu de fază II și, dacă frecvența ADR-urilor din studiul de fază II a fost mai mare decât cea înregistrată în studiul de fază III, evenimentul este inclus în RCP european la frecvența observată în studiul de fază II.
Algoritm aplicat pentru sindroame mielodisplazice:
• Studiu de fază III în sindroame mielodisplazice (populație de siguranță dublu-orb, diferență între lenalidomidă 5/10 mg și placebo pentru schema de dozare inițială cu apariție la cel puțin 2 subiecți))
o Toate evenimentele adverse apărute în timpul tratamentului la ≥ 5% dintre subiecții tratați cu lenalidomidă și o diferență de cel puțin 2% în procentul dintre lenalidomidă și placebo
o Toate evenimentele adverse de gradul 3 sau 4 care au apărut în timpul tratamentului la 1% dintre subiecții tratați cu lenalidomidă și o diferență de cel puțin 1% în procentul dintre lenalidomidă și placebo
o Toate evenimentele adverse grave care au apărut în timpul tratamentului la 1% dintre subiecții tratați cu lenalidomidă și o diferență de cel puțin 1% în procentul dintre lenalidomidă și placebo
• Studiu de fază II asupra sindroamelor mielodisplazice
o Toate evenimentele adverse care apar în timpul tratamentului la ≥ 5% dintre subiecții tratați cu lenalidomidă. Toate evenimentele adverse de gradul 3 sau 4 care apar în timpul tratamentului la 1% dintre subiecții tratați cu lenalidomidă. lenalidomidă
Tabelul 3: Reacții adverse raportate în studiile clinice la pacienții cu limfom cu celule de manta tratați cu lenalidomidă
^ Vezi secțiunea 4.8 Descrierea reacțiilor adverse selectate
♦ Evenimente adverse observate ca fiind grave în studiile clinice cu limfom cu celule de manta.
Algoritm aplicat limfomului cu celule de manta:
• Studiu de fază II controlat asupra limfomului cu celule de manta
o Toate evenimentele adverse apărute în timpul tratamentului la ≥ 5% dintre subiecții din brațul cu lenalidomidă și o diferență de cel puțin 2% în procentul dintre lenalidomidă și brațul de control
o Toate evenimentele adverse de gradul 3 sau 4 care au apărut în timpul tratamentului la ≥ 1% din subiecții din brațul cu lenalidomidă și o diferență de cel puțin 1,0% în procentul dintre lenalidomidă și brațul de control
o Toate evenimentele adverse grave care apar în timpul tratamentului la ≥ 1% dintre subiecții din brațul cu lenalidomidă și o diferență de cel puțin 1,0% în procentul dintre lenalidomidă și brațul de control
• Studiu de fază II cu un singur braț în limfomul cu celule de manta
o Toate evenimentele adverse apărute în timpul tratamentului la ≥ 5% dintre subiecți
o Toate evenimentele adverse de gradul 3 sau 4 care au apărut în timpul tratamentului raportate la 2 sau mai mulți subiecți
Tabel rezumativ al reacțiilor adverse după punerea pe piață
În plus față de reacțiile adverse menționate mai sus identificate din studiile clinice esențiale, următorul tabel a fost compilat pe baza datelor colectate după punerea pe piață.
Tabelul 4: Reacții adverse raportate la utilizarea după punerea pe piață la pacienții tratați cu lenalidomidă
^ Vezi secțiunea 4.8 Descrierea reacțiilor adverse selectate
Descrierea reacțiilor adverse selectate
Teratogenitate
Lenalidomida este legată structural de talidomidă, o substanță activă cu efect teratogen cunoscut la om, care provoacă defecte congenitale grave care pun viața în pericol. Lenalidomida a indus malformații la maimuțe similare celor descrise pentru talidomidă (vezi secțiunile 4.6 și 5.3) .un efect teratogen al lenalidomidei este de așteptat la om în timpul sarcinii.
Neutropenie și trombocitopenie
• Pacienți nou diagnosticați cu mielom multiplu tratați cu lenalidomidă în asociere cu doză mică de dexametazona
La pacienții cu mielom multiplu nou diagnosticat, combinația de lenalidomidă cu doză mică de dexametazonă este asociată cu o incidență redusă a neutropeniei de gradul 4 (8,5% în Rd și Rd18, comparativ cu 15% în MPT). Neutropenia febrilă Gradul 4 a fost observată rar (0,6%, comparativ cu 0,7% în MPT).
La pacienții cu mielom multiplu nou diagnosticat, combinația de lenalidomidă cu doză mică de dexametazonă este asociată cu o incidență redusă a trombocitopeniei de gradul 3 și 4 (8,1% în Rd și Rd18, comparativ cu 11% în MPT).
• Pacienți nou diagnosticați cu mielom multiplu tratați cu lenalidomidă în asociere cu melfalan și prednison
La pacienții cu mielom multiplu nou diagnosticat, combinația de lenalidomidă cu melfalan și prednison este asociată cu o incidență mai mare a neutropeniei de gradul 4 (34,1% în MMR + R / MPR + p, comparativ cu 7,8% în MPp + p) O incidență mai mare a neutropeniei febrile de gradul 4 a fost observată (1,7% în MPR + R / MPR + p, comparativ cu 0,0% în MPp + p).
La pacienții cu mielom multiplu nou diagnosticat, combinația de lenalidomidă cu melfalan și prednison este asociată cu o incidență mai mare a trombocitopeniei de gradul 3 și gradul 4 (40,4% la pacienții tratați cu MMR + R / MMR + p, comparativ cu 13,7% la pacienți tratat cu MPp + p).
• Mielom multiplu cu cel puțin o terapie anterioară
La pacienții cu mielom multiplu, combinația de lenalidomidă și dexametazonă este asociată cu o incidență mai mare a neutropeniei de gradul 4 (5,1% dintre pacienții tratați cu lenalidomidă / dexametazonă versus 0,6% dintre pacienții tratați cu placebo / dexametazonă Au fost observate rareori episoade de neutropenie febrilă de gradul 4) la 0,6% dintre pacienții tratați cu lenalidomidă / dexametazonă versus 0,0% dintre pacienții tratați cu placebo / dexametazonă).
La pacienții cu mielom multiplu, combinația de lenalidomidă și dexametazonă este asociată cu o incidență mai mare a trombocitopeniei de gradul 3 și gradul 4 (la 9,9% și 1,4% dintre pacienții tratați cu lenalidomidă / dexametazonă, comparativ cu 2,3% și 0,0% din pacienți tratați cu placebo / dexametazonă).
• Sindroame mielodisplazice
La pacienții cu sindroame mielodisplazice, lenalidomida este asociată cu o incidență mai mare a neutropeniei de gradul 3 sau 4 (74,6% dintre pacienții tratați cu lenalidomidă versus 14,9% dintre pacienții tratați cu placebo în studiul de fază III). Episoadele de neutropenie febrilă de gradul 3 sau 4 au fost observate la 2,2% dintre pacienții tratați cu lenalidomidă, comparativ cu 0,0% dintre pacienții tratați cu placebo. Lenalidomida este asociată cu o incidență mai mare a trombocitopeniei de gradul 3 sau 4 (37% la pacienții tratați cu lenalidomidă și 1,5% la pacienții tratați cu placebo în studiul de fază III).
• Limfom cu celule de manta
La pacienții cu limfom cu celule de manta, lenalidomida este asociată cu o incidență mai mare a neutropeniei de gradul 3 sau 4 (43,7% dintre pacienții tratați cu lenalidomidă comparativ cu 33,7% dintre pacienții din brațul de control în studiul de fază III). Au fost observate episoade de neutropenie febrilă de gradul 3 sau 4 la 6,0% dintre pacienții tratați cu lenalidomidă, comparativ cu 2,4% dintre pacienții din brațul de control.
Tromboembolism venos
Un risc crescut de tromboză venoasă profundă (TVP) și embolie pulmonară (PE) este asociat cu utilizarea lenalidomidei și a dexametazonei la pacienții cu mielom multiplu și, într-o măsură mai mică, la pacienții tratați cu melfalan și prednison sau ca monoterapie la pacienții cu sindroame. limfom mielodisplazic și celular de manta tratat cu lenalidomidă (vezi pct. 4.5) La acești pacienți, administrarea concomitentă de agenți eritropoietici sau antecedente de TVP pot crește, de asemenea, riscul de tromboză.
Infarct miocardic
Au fost observate cazuri de infarct miocardic la pacienții cărora li sa administrat lenalidomidă, în special la cei cu factori de risc cunoscuți.
Tulburări de sângerare
Tulburările de sângerare sunt enumerate în diferite clasificări pe baza organului implicat: patologii ale sângelui și ale sistemului limfatic; patologii ale sistemului nervos (hemoragie intracraniană); patologii respiratorii, toracice și mediastinale (epistaxis); hemoragie rectală); patologii renale și urinare (hematurie); traume, otrăviri și complicații procedurale (contuzie); patologii vasculare (echimoză).
Reactii alergice
Au fost raportate cazuri de reacții alergice / de hipersensibilitate. În literatura de specialitate a fost raportată o posibilă reacție încrucișată între lenalidomidă și talidomidă.
Reacții cutanate severe
Au fost raportate cazuri de SSJ și NTE. Pacienții cu antecedente de erupții cutanate severe asociate tratamentului cu talidomidă nu trebuie să primească lenalidomidă.
A doua tumori primare
* În studiile clinice la pacienții cu mielom tratați anterior cu lenalidomidă / dexametazonă versus martori, constând în principal din cancer de piele cu celule bazale sau cu celule scuamoase.
Leucemie mieloidă acută
• Mielom multiplu
În studiile clinice efectuate pe mielom multiplu nou diagnosticat, au fost observate cazuri de LAM la pacienții tratați cu lenalidomidă în asociere cu melfalan sau la scurt timp după doze mari de melfalan și ASCT (vezi pct. 4.4). Această creștere nu a fost observată în studiile clinice la pacienții nou diagnosticați cu mielom multiplu tratați cu lenalidomidă în asociere cu doze mici de dexametazonă, comparativ cu talidomida în combinație cu melfalan și prednison.
• Sindroame mielodisplazice
Variabilele de bază, inclusiv anomalii citogenetice complexe și mutația TP53, sunt asociate cu progresia la LMA la subiecții dependenți de transfuzie cu anomalie de deleție 5q izolată (vezi pct. 4.4). Riscul cumulativ de progresie la LMA estimat la 2 ani a fost de 13,8% la pacienții cu anomalie de deleție 5q izolată, comparativ cu 17,3% la pacienții cu anomalie de deleție 5q izolată și o „anomalie citogenetică suplimentară. Și 38,6% la pacienții cu cariotip complex.
Într-o „analiză post-hoc a unui studiu clinic efectuat cu Revlimid în sindroame mielodisplazice, rata estimată de progresie pe 2 ani către LMA a fost de 27,5% la pacienții cu IHC-p53-pozitivi și de 3,6% la pacienții cu IHC-p53-pozitivi. IHC- p53-negativ (p = 0,0038). La pacienții cu IHC-p53-pozitiv, s-a observat o rată mai mică de progresie la LMA în rândul celor care au obținut răspuns de independență transfuzională (11,1%), comparativ cu cei care nu au răspuns (34,8%).
Tulburări hepatice
Au fost observate următoarele reacții adverse după punerea pe piață (frecvență necunoscută): insuficiență hepatică acută și colestază (ambele punând viața în pericol), hepatită toxică, hepatită citolitică, hepatită citolitică mixtă / colestatică.
Rabdomioliza
Au fost observate cazuri rare de rabdomioliză, unele dintre ele când lenalidomida a fost administrată cu o statină.
Tulburări ale glandei tiroide
Au fost observate cazuri de hipotiroidism și hipertiroidism (vezi pct. 4.4 Tulburări tiroidiene).
Reacția de erupție tumorală și sindromul de liză tumorală
În studiul MCL-002, aproximativ 10% dintre pacienții tratați cu lenalidomidă au prezentat TFR, comparativ cu 0% în brațul de control. Cele mai multe evenimente au avut loc în ciclul 1, toate au fost considerate legate de tratament și cele mai multe rapoarte au fost de gradul 1 sau 2. Pacienții cu MIPI ridicat la diagnostic și boala caracterizată prin mase tumorale mari (cel puțin o leziune cu diametrul cel mai lung de ≥ 7 cm ) la momentul inițial poate prezenta un risc pentru TFR. În studiul MCL-002, TLS a fost raportat pentru un pacient din fiecare dintre cele două brațe de tratament. În studiul de sprijin MCL-001, aproximativ 10% dintre subiecți au prezentat TFR; toate rapoartele au fost de gradul 1 sau 2 ca severitate și toate au fost considerate legate de tratament. Cele mai multe evenimente au avut loc în ciclul 1. Nu au fost raportate TLS în studiul MCL-001 (vezi pct. 4.4).
Tulburări gastrointestinale
În timpul tratamentului cu lenalidomidă au fost observate perforații gastrointestinale, care pot duce la complicații septice și pot fi asociate cu un rezultat fatal.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate care apar după autorizarea medicamentului este importantă, deoarece permite monitorizarea continuă a raportului beneficiu / risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacții adverse suspectate prin intermediul sistemului național de raportare. În „Anexa V .
04.9 Supradozaj -
Nu există experiență specifică în tratamentul supradozajului cu lenalidomidă la pacienți, deși în studiile de stabilire a dozelor unii pacienți au fost expuși la doze de până la 150 mg și, în studiile cu doză unică, unii pacienți au fost expuși la doze de până la 400 mg. În aceste studii, toxicitatea care limitează doza a fost în esență de natură hematologică. În caz de supradozaj, se recomandă terapia de susținere.
05.0 PROPRIETĂȚI FARMACOLOGICE -
05.1 "Proprietăți farmacodinamice -
Grupa farmacoterapeutică: alte imunosupresoare.
Codul ATC: L04 AX04.
Mecanism de acțiune
Mecanismul de acțiune al lenalidomidei include proprietăți anti-neoplazice, anti-angiogene, pro-eritropoietice și imunomodulatoare. În mod specific, lenalidomida inhibă proliferarea celulelor tumorale hematopoietice specifice (inclusiv celulele plasmatice canceroase ale MM și cele cu deleție a cromozomului 5). îmbunătățește imunitatea mediată de celulele T și celulele natural killer (NK) și crește numărul de celule NKT; inhibă angiogeneza prin blocarea migrației și aderenței celulelor endoteliale și formarea de microvase; crește producția de hemoglobină fetală de către celulele stem hematopoietice CD34 + și inhibă producția de citokine proinflamatorii (de exemplu TNF-α și IL-6) de către monocite.
În MDS cu anomalie de deleție 5q izolată, s-a demonstrat că lenalidomida inhibă selectiv clona anormală, crescând apoptoza celulelor Del (5q).
Lenalidomida se leagă direct de cereblon, o componentă a unui complex enzimatic cullin-RING E3 ubiquitin ligază, care include proteina 1 de legare a deteriorării acidului dezoxiribonucleic (ADN),Proteina de legare a deteriorării ADN-1), cullina 4 (CUL4) și regulatorul de tăiere 1 (Roc1). În prezența lenalidomidei, cereblonul se leagă de proteinele de substrat Aiolos și Ikaros, care sunt factori de transcripție limfoidă, provocând ubiquitinarea și degradarea ulterioară a acestora, cu consecința efectelor citotoxice și imunomodulatoare.
Eficacitate și siguranță clinică
Lenalidomida a fost evaluată în două studii de fază III în mielomul multiplu nou diagnosticat și în două studii de fază III în mielomul multiplu refractar recidivant, așa cum este descris mai jos.
Pe mine Și L sau m un multiplu nou diagnosticat
Lenalidomida în asociere cu dexametazonă la pacienții care nu sunt eligibili pentru transplant de celule stem
Eficacitatea și siguranța lenalidomidei au fost evaluate într-un studiu de fază III, multicentric, randomizat, deschis, cu trei brațe (MM-020) la pacienți cu vârsta de 65 de ani sau peste sau, dacă au vârsta sub 65 de ani, care au fost neeligibili pentru transplantul de celule stem din cauza deciziei pacientului sau a indisponibilității transplantului de celule stem din costuri sau din alte motive. Studiul (MM-020) a comparat lenalidomida și dexametazona (Rd) administrate pentru 2 durate de tratament diferite (de exemplu până la progresia bolii [brațul Rd] sau până la optsprezece cicluri de 28 de zile [72 săptămâni, brațul Rd18]) cu melfalan, prednison și talidomidă (MPT) timp de până la douăsprezece cicluri de 42 de zile (72 de săptămâni). Pacienții au fost randomizați (1: 1: 1) la unul dintre cele 3 brațe de tratament. La randomizare, pacienții au fost stratificați în funcție de vârstă (≤ 75 ani față de> 75 ani), stadiu (ISS stadiile I și II față de stadiul III) și țară.
Pacienții din brațele Rd și Rd18 au primit lenalidomidă 25 mg o dată pe zi în zilele 1 - 21 din ciclurile de 28 de zile, conform brațului protocol. Dexametazona 40 mg a fost administrată o dată pe zi în zilele 1, 8, 15 și 22 din fiecare ciclu de tratament de 28 de zile. Doza inițială și regimul pentru Rd și Rd18 au fost ajustate în funcție de vârstă și funcția renală (vezi pct. 4.2). Pacienții cu vârsta> 75 de ani au primit o doză de dexametazonă de 20 mg o dată pe zi în zilele 1, 8, 15 și 22 din fiecare 28 de zile ciclu de tratament Toți pacienții au suferit profilaxie anticoagulantă (heparină cu greutate moleculară mică, warfarină, heparină, aspirină cu doză mică) în timpul studiului.
Obiectivul principal al eficacității în studiu a fost supraviețuirea fără progresie (Progresie Supraviețuire gratuită, PFS). În total, 1623 pacienți au fost înrolați în studiu: 535 pacienți randomizați la Rd, 541 pacienți randomizați la Rd18 și 547 pacienți randomizați la MPT. Caracteristicile demografice și legate de boală ale pacienților la momentul inițial au fost bine echilibrate în toate cele 3 brațe.În general, subiecții studiați aveau boală avansată: din totalul populației studiate, 41% erau în stadiul III al ISS, 9% aveau insuficiență renală severă (clearance-ul creatininei [CLcr]
Într-o analiză actualizată a PFS, PFS2 și a supraviețuirii globale (OS) folosind data limită de 3 martie 2014, unde timpul mediu de urmărire pentru toți subiecții supraviețuitori a fost de 45,5 luni, rezultatele studiului sunt prezentate în Tabelul 5.
Tabelul 5: Rezumatul datelor generale de eficacitate
AMT = terapie anti-mielom; CI = interval de încredere; CR = răspuns complet; d = doză mică de dexametazonă; HR = raportul de pericol; IMWG = Grupul de lucru internațional pentru mielom; IRAC = Comitetul independent de soluționare a răspunsului; M = melfalan; max = maxim; min = minim; NS = nu este estimabil; OS = supraviețuirea generală; P = prednison; PFS = supraviețuire fără progresie; PR = răspuns parțial; R = lenalidomidă; Rd = Rd administrat până la progresia documentată a bolii; Rd18 = Rd administrat ≥ 18 cicluri; SE = eroare standard; T = talidomidă; VGPR = răspuns parțial optim; vs = versus.
a Mediana se bazează pe estimarea Kaplan-Meier.
b IC 95% despre mediana.
c Pe baza modelului de pericole proporționale Cox, care compară funcțiile de pericol asociate cu brațele de tratament indicate.
d Valoarea p se bazează pe testul de rang logaritmic nestratificat al diferențelor în curbele Kaplan-Meier între brațele de tratament indicate.
e Endpoint explorator (PFS2)
f Mediana este statistica univariată fără corecție a trunchierii.
g Evaluarea îmbunătățită a răspunsului evaluat în timpul fazei de tratament a studiului (pentru definițiile fiecărei categorii de răspuns Data limită a datelor = 24 mai 2013).
h Data limită = 24 mai 2013
Lenalidomidă în asociere cu melfalan și prednison, urmată de monoterapie de întreținere, în pacienții care nu sunt eligibili pentru transplant
Siguranța și eficacitatea lenalidomidei (MPR) au fost evaluate într-un studiu de fază III multicentric, randomizat, dublu-orb, cu 3 brațe (MM-015) la pacienți cu vârsta de 65 de ani și peste și cu creatinină serică. 75 de ani) și stadiu (ISS etapele I și II vs etapa III).
Acest studiu a examinat utilizarea terapiei combinate MMR (melphalan 0,18 mg / kg pe cale orală în zilele 1-4 din ciclurile repetate de 28 de zile; prednison 2 mg / kg pe cale orală în zilele 1-4 din ciclurile repetate de 28 de zile; și lenalidomida 10 mg / zi pe cale orală în zilele 1-21 ale ciclurilor repetate de 28 de zile), pentru terapia de inducție, până la maximum 9 cicluri. monoterapie de întreținere, a început cu lenalidomidă 10 mg pe cale orală în zilele 1-21 ale ciclurilor repetate de 28 de zile, până la progresia bolii
Obiectivul principal al eficacității în studiu a fost supraviețuirea fără progresie (SFP). În total, 459 pacienți au fost înrolați în studiu: 152 pacienți randomizați la MMR + R, 153 pacienți randomizați la MMR + p și 154 pacienți randomizați la MPp + p Caracteristicile demografice și legate de boală ale pacienților la momentul inițial au fost bine echilibrate în toate cele 3 brațe; în special, aproximativ 50% dintre pacienții înrolați în fiecare braț au prezentat următoarele caracteristici: stadiul III al ISS și clearance-ul creatininei.
Într-o „analiză a PFS, PFS2 și OS utilizând data limită din aprilie 2013, pentru care timpul mediu de urmărire pentru toți subiecții supraviețuitori a fost de 62,4 luni, rezultatele studiului sunt prezentate în Tabelul 6.
Tabelul 6: Rezumatul datelor generale de eficacitate
CI = interval de încredere; CR = răspuns complet; HR = raportul de pericol; M = melfalan; NS = nu este estimabil; OS = supraviețuirea generală; p = placebo; P = prednison;
PD = boală progresivă; PR = răspuns parțial; R = lenalidomidă; SD = boală stabilă; VGPR = răspuns parțial optim.
a Mediana se bazează pe estimarea Kaplan-Meier.
¤ PFS2 (un obiectiv final explorator) a fost definit pentru toți pacienții (ITT) ca timpul de la randomizare la inițierea terapiei anti-mielom de linia a treia sau decesul din orice cauză, pentru toți pacienții randomizați
Studii de susținere a mielomului multiplu nou diagnosticat
Un studiu deschis, randomizat, multicentric de fază III (ECOG E4A03) a fost efectuat la 445 de pacienți cu mielom multiplu nou diagnosticat; 222 de pacienți au fost randomizați la brațul cu lenalidomidă / doză mică de dexametazonă și 223 au fost randomizați la brațul cu lenalidomidă / doză standard de dexametazonă. Pacienții randomizați în brațul cu lenalidomidă / doză standard de dexametazonă au primit lenalidomidă 25 mg / zi în zilele 1 până la 21, la fiecare 28 de zile, plus dexametazonă 40 mg / zi în zilele 1 până la 4, 9 până la 12 și 17 la 20, la fiecare 28 de zile, pentru primele patru cicluri. Pacienții randomizați la brațul cu lenalidomidă / doză mică de dexametazonă au primit lenalidomidă 25 mg / zi în zilele 1 până la 21, la fiecare 28 de zile, plus doze mici de dexametazonă 40 mg / zi în zilele 1, 8, 15 și 22, la fiecare 28 de zile . În grupul cu lenalidomidă / doză mică de dexametazonă, 20 de pacienți (9,1%) au prezentat cel puțin o întrerupere a dozei, comparativ cu 65 de pacienți (29,3%) din brațul cu lenalidomidă / doză standard de dexametazonă.
Într-o analiză post-hoc, cea mai mică mortalitate a fost observată la brațul cu lenalidomidă / doză mică de dexametazonă 6,8% (15/220), comparativ cu brațul cu lenalidomidă / doză standard de dexametazonă 19,3% (43/223), la numărul nou diagnosticat populație de pacienți cu mielom, cu o urmărire mediană de 72,3 săptămâni.
Cu toate acestea, cu o monitorizare extinsă, diferența de supraviețuire globală în favoarea dozei mici de lenalidomidă / dexametazonă tinde să scadă.
Mielom multiplu cu cel puțin o terapie anterioară
Eficacitatea și siguranța lenalidomidei au fost evaluate în două studii de grup paralel (MM-009 și MM-010) multicentric, randomizat, dublu-orb, controlat cu placebo (MM-009 și MM-010) de fază III, în asociere cu dexametazonă versus monoterapie cu dexametazonă la pacienții tratați anterior cu mielom multiplu. Dintre cei 353 de pacienți incluși în studiile MM-009 și MM-010 tratați cu lenalidomidă / dexametazonă, 45,6% au avut vârsta de 65 de ani sau mai mult. Dintre cei 704 de pacienți evaluați în studiile MM-009 și MM-010, 44,6% aveau 65 de ani sau mai mult.
În ambele studii, pacienții din grupul de lenalidomidă / dexametazonă (len / des) au primit lenalidomidă 25 mg pe cale orală o dată pe zi în zilele 1 până la 21 și o capsulă placebo cu aspect identic o dată pe zi. grupul placebo / dexametazonă (placebo / des) a luat 1 capsulă de placebo în zilele 1-28 ale fiecărui ciclu de 28 de zile. Pacienții din ambele grupuri au luat 40 mg de dexametazonă pe cale orală o dată pe zi în zilele 1-4, 9-12 și
17-20 din fiecare ciclu de 28 de zile, pentru primele 4 cicluri de terapie. După primele 4 cure de terapie, doza de dexametazonă a fost redusă la 40 mg pe cale orală o dată pe zi, în zilele 1-4 din fiecare ciclu de 28 de zile. În ambele studii, tratamentul urma să continue până la progresia bolii. Ajustările dozelor au fost permise în ambele studii. pe baza constatărilor clinice și de laborator.
Obiectivul principal de eficacitate în ambele studii a fost timpul până la progresia bolii (TTP, timpul până la progresie). Un total de 353 de pacienți au fost evaluați în studiul MM-009: 177 în grupul cu lenalidomidă / dexametazonă și 176 în grupul placebo / dexametazonă. Un total de 351 de pacienți au fost evaluați în studiul MM-010: 176 în grupul cu lenalidomidă / dexametazonă și 175 în grupul placebo / dexametazonă.
În ambele studii, grupurile de lenalidomidă / dexametazonă și placebo / dexametazonă au avut caracteristici demografice inițiale comparabile și caracteristici legate de boală. Ambele populații de pacienți au avut o vârstă mediană de 63 de ani, cu un raport comparabil între pacienții bărbați și femei.Grupul de Oncologie Cooperativă din Est), atât numărul, cât și tipul terapiilor anterioare au fost comparabile în ambele grupuri.
Analize interimar pre-programate pentru ambele studii au arătat că terapia combinată cu lenalidomidă / dexametazonă a arătat o îmbunătățire semnificativă statistic (p
O analiză extinsă a eficacității de urmărire a fost efectuată pentru o medie de 130,7 săptămâni. Tabelul 7 prezintă rezultatele analizelor de eficacitate de urmărire - studii comune MM-009 și MM-010.
În această analiză de urmărire extinsă, media TTP a fost de 60,1 săptămâni (IÎ 95%: 44,3, 73,1) la pacienții tratați cu lenalidomidă / dexametazonă (N = 353) comparativ cu mediana de 20, 1 săptămână (IÎ 95%: 17,7, 20,3) la pacienții tratați cu placebo / dexametazonă (N = 351). Supraviețuirea mediană fără progresie a fost de 48,1 săptămâni (IÎ 95%: 36,4, 62,1) la pacienții tratați cu lenalidomidă / dexametazonă comparativ cu un timp median de 20,0 săptămâni (IÎ 95%: 16, 1, 20,1) la pacienții tratați cu placebo / dexametazonă . Durata medie a tratamentului a fost de 44,0 săptămâni (min: 0,1, maxim: 254,9) pentru lenalidomidă / dexametazonă și 23,1 săptămâni (min: 0,3, maxim: 238,1) pentru placebo / dexametazonă. În ambele studii, ratele de răspuns complete (CR, răspuns complet), răspuns parțial (PR, răspuns parțial) și răspunsul general (CR + PR) în grupul cu lenalidomidă / dexametazonă au rămas semnificativ mai mari decât în grupul cu dexametazonă / placebo. Supraviețuirea globală mediană în analiza extinsă de urmărire a studiilor comune a fost de 164,3 săptămâni (IÎ 95%: 145,1, 192,6) la pacienții tratați cu lenalidomidă / dexametazonă comparativ cu 136,4 săptămâni (IÎ 95%: 113,1, 161,7) la pacienții tratați cu În ciuda faptului că 170 din cei 351 de pacienți randomizați la tratament placebo / dexametazonă au primit tratament cu lenalidomidă după progresia bolii sau după orbire, analiza globală a supraviețuirii a demonstrat un avantaj de supraviețuire semnificativ statistic pentru grupul cu lenalidomidă / dexametazonă comparativ cu placebo / grup de dexametazona (grad de periculozitate = 0,833, 95% CI = [0,687, 1,009], p = 0,045).
Tabelul 7: Rezumatul rezultatelor analizelor de eficacitate la data limită pentru urmărirea extinsă - Studii comune MM-009 și MM-010 (date limită respective: 23 iulie 2008 și 2 martie 2008)
a: Analiza univariantă pe două fețe care compară curbele de supraviețuire între grupurile de tratament. b: Test chi-pătrat cu două cozi cu corecție de continuitate.
Sindroame mielodisplazice
Eficacitatea și siguranța lenalidomidei au fost evaluate la pacienții cu anemie dependentă de transfuzie din cauza sindroamelor mielodisplazice cu risc scăzut sau intermediar 1 asociate cu anomalie citogenetică cu deleție 5q, cu sau fără alte anomalii citogenetice, în două studii principale: o fază III, multicentrică. , randomizat, dublu-orb, controlat cu placebo, cu 3 brațe, cu două doze de lenalidomidă orală (10 mg și 5 mg) versus studiu placebo (MDS-004); și un studiu de fază II, multicentric, cu un singur braț, deschis, pe lenalidomidă (10 mg) (MDS-003).
Rezultatele de mai jos reprezintă populația intenționată de tratat studiată în MDS-003 și MDS-004; rezultatele pentru subpopulația cu ștergere izolată 5q sunt prezentate separat (a se vedea secțiunea 4.1 pentru indicația aprobată).
În studiul MDS-004, în care 205 pacienți au fost randomizați în mod egal la tratament cu lenalidomidă 10 mg, 5 mg sau placebo, analiza eficacității principale a constat în compararea ratelor de răspuns la independența transfuziei în brațele de 10 mg lenalidomidă și 5 mg comparativ cu placebo braț (fază dublu-orb de la 16 la 52 de săptămâni și fază deschisă până la un total de 156 de săptămâni). La pacienții care nu au prezentat cel puțin un răspuns eritroid ușor după 16 săptămâni, tratamentul a fost oprit. cel puțin un răspuns eritroid ușor ar putea continua terapia până la recurența eritroidiană, progresia bolii sau toxicitate inacceptabilă. După 16 săptămâni de tratament li s-a permis trecerea de la placebo la 5 mg de lenalidomidă sau continuarea tratament cu lenalidomidă la o doză mai mare (5 mg până la 10 mg).
În studiul MDS-003, în care 148 de pacienți au fost tratați cu lenalidomidă în doză de 10 mg, analiza principală a eficacității a constat într-o evaluare a eficacității tratamentelor cu lenalidomidă în realizarea ameliorării hematopoietice la subiecții cu sindroame mielodisplazice. 1.
Tabelul 8: Rezumatul rezultatelor de eficacitate - MDS-004 (faza dublu-orb) și MDS-003, populație intenționată de tratat
† Subiecții tratați cu lenalidomidă 10 mg pe 21 din ciclul de 28 de zile
†† Subiecții tratați cu lenalidomidă 5 mg la 28 din ciclul de 28 de zile
* Majoritatea pacienților din grupul placebo au întrerupt tratamentul dublu-orb din cauza lipsei de eficacitate după 16 săptămâni de tratament, înainte de intrarea în faza deschisă
# Asociat cu o creștere a Hgb de ≥ 1 g / dL
∞ Nu a fost atins (mediana nu a fost atinsă)
În studiul MDS-004, o proporție semnificativ mai mare de pacienți cu sindroame mielodisplazice a atins obiectivul primar al independenței transfuziei (> 182 zile) cu lenalidomidă 10 mg, comparativ cu placebo (55,1% față de 6,0%) Dintre cei 47 de pacienți cu anomalie citogenetică .
Del (5q) izolat și tratat cu 10 mg lenalidomidă, 27 de pacienți (57,4%) au obținut independență de transfuziile de eritrocite.
Timpul mediu până la independența transfuziei în brațul de 10 mg lenalidomidă a fost de 4,6 săptămâni. Durata medie a independenței transfuziei nu a fost atinsă în niciunul dintre brațele de tratament, dar se așteaptă să depășească 2 ani pentru subiecții tratați. Cu lenalidomidă. hemoglobina (Hgb) de la momentul inițial în brațul de 10 mg a fost de 6,4 g / dL.
Obiectivele suplimentare ale studiului au inclus răspunsul citogenetic (răspunsurile citogenetice majore și minore au fost observate la 30,0% și 24,0% dintre subiecții din brațul de 10 mg, respectiv), evaluarea calității vieții legate de sănătate (HRQoL) și progresia către leucemie mieloidă acută. Rezultatele răspunsului citogenetic și HRQoL au fost în concordanță cu rezultatele obiectivului primar și în favoarea tratamentului cu lenalidomidă față de placebo.
În studiul MDS-003, un procent ridicat de pacienți cu sindroame mielodisplazice au obținut independență transfuzională (> 182 zile) cu lenalidomidă 10 mg (58,1%). Timpul mediu până la independența transfuziei a fost de 4,1 săptămâni. Durata medie a independenței transfuziei a fost de 114,4 săptămâni. Creșterea mediană a hemoglobinei (Hgb) a fost de 5,6 g / dl.
Răspunsurile citogenetice majore și minore au fost observate la 40,9% și, respectiv, 30,7% dintre subiecți.
Un procent mare de subiecți înrolați în MDS-003 (72,9%) și MDS-004 (52,7%) au fost tratați anterior cu agenți de stimulare a eritropoiezei.
Limfom cu celule de manta
Eficacitatea și siguranța lenalidomidei la pacienții cu limfom cu celule de manta au fost evaluate într-un studiu de fază II, multicentric, randomizat, deschis, comparativ cu alegerea de către investigator a monoterapiei la pacienții refractari la ultimul regim sau care au prezentat de la una până la trei recăderi ( Studiați MCL-002).
Au fost înscriși pacienți cu vârsta de cel puțin 18 ani cu limfom cu celule de manta confirmat histologic și boală măsurabilă la CT. Pacienții trebuiau să primească un tratament anterior adecvat cu cel puțin un regim de chimioterapie asociat anterior. În plus, pacienții au fost nevoiți să nu fie eligibili pentru chimioterapie intensivă și / sau transplant la includerea în studiu. Pacienții au fost randomizați 2: 1 la lenalidomidă sau brațul de control. Alegerea tratamentului investigatorului a fost decisă mai devreme. citarabină, rituximab, fludarabină sau gemcitabină.
Lenalidomida a fost administrată pe cale orală, la o doză de 25 mg o dată pe zi, în primele 21 de zile (G1 până la G21) de cicluri repetate de 28 de zile până la progresie sau toxicitate inacceptabilă. Pacienții cu insuficiență renală moderată urmau să primească o doză inițială mai mică de lenalidomidă (10 mg pe zi) cu același program.
Demografia de bază a fost comparabilă între lenalidomidă și brațele de control. Ambele populații de pacienți au avut o vârstă mediană de 68,5 ani, cu un raport comparabil dintre pacienții de sex masculin și de sex feminin.
Obiectivul principal de eficacitate în MCL-002 a fost supraviețuirea fără progresie (PFS).
Rezultatele eficacității pentru populația cu intenție de tratare (ITT) au fost evaluate de către Comisia de revizuire independentă (Comitetul independent de revizuire, IRC) și sunt prezentate în tabelul de mai jos.
Tabelul 9: Rezumatul rezultatelor de eficacitate - studiu MCL-002, populație intenționată de tratat
CI = interval de încredere; CRR = rata de răspuns completă; CR = răspuns complet; CRu = răspunsul complet nu este confirmat; DMC = comitetul de monitorizare a datelor; ITT = intenția de a trata; HR = raportul de pericol; KM = Kaplan-Meier; MIPI = Mantle Cell Lymphoma International Prognostic Index; NP = nu este relevant; ORR = rata generală de răspuns; PD = boală progresivă; PFS = supraviețuire fără progresie; PR = răspuns parțial; SCT = transplant de celule stem; SD = boală stabilă; SE = eroare standard.
a Mediana se bazează pe estimarea KM.
b Intervalul a fost calculat ca IC 95% în raport cu timpul mediu de supraviețuire.
e Media și mediana sunt statistici univariate fără corecție a trunchierii.
d Variabilele de stratificare au inclus timpul de la diagnostic la prima doză (
e Testarea secvențială s-a bazat pe o medie ponderată a statisticii testului rangului jurnalului, utilizând testul de rang jurnal netratificat pentru creșterea mărimii eșantionului și testul de rang jurnal nestratificat din analiza primară. Ponderile se bazează pe evenimentele observate la data celei de-a treia întâlniri a RCD și se bazează pe diferența dintre evenimentele observate și evenimentele așteptate la momentul analizei primare.
Sunt prezentate HR secvențiale asociate și CI 95% corespunzătoare.
În studiul MCL-002 la populația ITT, s-a înregistrat o creștere globală aparentă a deceselor în 20 de săptămâni la brațul cu lenalidomidă, 22/170 (13%), comparativ cu 6/84 (7%) la brațul de control. La pacienții cu sarcină tumorală mare, cifrele corespunzătoare au fost 16/81 (20%) și 2/28 (7%) (vezi pct. 4.4).
Populația pediatrică
Agenția Europeană a Medicamentului a renunțat la obligația de a prezenta rezultatele studiilor cu Revlimid la toate subseturile populației pediatrice pentru mielom multiplu, sindroame mielodisplazice și limfom cu celule de manta (a se vedea secțiunea 4.2 pentru informații privind „utilizarea pediatrică).
05.2 "Proprietăți farmacocinetice -
Lenalidomida are un atom de carbon asimetric; de aceea molecula sa există în formele optic active S (-) și R (+). Lenalidomida este produsă ca un amestec racemic. Lenalidomida este în general mai solubilă în solvenți organici, dar prezintă o solubilitate maximă în soluție de HCI 0,1N.
Absorbţie
Lenalidomida se absoarbe rapid după administrarea orală la voluntari sănătoși, în condiții de repaus alimentar, atingând concentrații plasmatice maxime între 0,5 și 2 ore după administrare. La pacienți și voluntari sănătoși concentrația maximă (Cmax) și zona l "sub curba concentrație-timp ( ASC) cresc proporțional cu creșterea dozei. Dozele repetate nu determină acumularea semnificativă a medicamentului. În plasmă, concentrația relativă a enantiomerilor S- și R- a lenalidomidei este de aproximativ 56% și respectiv 44%.
Administrarea concomitentă a unei mese bogate în calorii și bogate în grăsimi la voluntarii sănătoși reduce gradul de absorbție, rezultând o scădere cu aproximativ 20% a zonei sub ASC și o scădere cu 50% a Cmax plasmatic. Cu toate acestea, în studiile de înregistrare a sindromului mielom multiplu și mielodisplazic în care au fost stabilite siguranța și eficacitatea lenalidomidei, medicamentul a fost administrat fără a lua în considerare aportul de alimente. Prin urmare, lenalidomida poate fi administrată cu sau fără alimente.
Analizele farmacocinetice populaționale indică faptul că rata de absorbție a lenalidomidei orale este similară la pacienții cu mielom multiplu, pacienții cu sindroame mielodisplazice și pacienții cu limfom cu celule de manta.
Distribuție
In vitro, Lenalidomida marcată cu 14C este slab legată de proteinele plasmatice, cu o valoare medie de 23% și respectiv 29% la pacienții cu mielom multiplu și voluntari sănătoși.
Lenalidomida este prezentă în material seminal (
Biotransformare și eliminare
Rezultatele studiilor de metabolism uman efectuate in vitro indică faptul că lenalidomida nu este metabolizată de enzimele citocromului P450, sugerând că administrarea de lenalidomidă împreună cu medicamente care inhibă enzimele citocromului P450 este puțin probabil să producă interacțiuni medicamentoase metabolice la om. in vitro indicați că lenalidomida nu are efect inhibitor asupra CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A sau UGT1A1. Prin urmare, este puțin probabil ca lenalidomida să provoace interacțiuni medicamentoase relevante clinic atunci când este administrată concomitent cu substraturi ale acestor enzime.
Educaţie in vitro indicați că lenalidomida nu este un substrat al proteinelor de rezistență la cancerul de sân uman (BCRP), transportoare de proteine multirezistență (MRP) MRP1, MRP2 sau MRP3, transportoare de anioni organici (OAT) OAT1 și OAT3, transportor de anioni organici polipeptidici (OATP) OATP1B1, transportori de cationi (OCT) OCT1 și OCT2, proteine de extrudare a medicamentelor și toxinelor (MATE) MATE1 și noi transportori de cationi organici (OCTN) OCTN1 și OCTN2.
Educaţie in vitro indicați că lenalidomida nu are efect inhibitor asupra pompei de export de sare biliară umană (BSEP), BCRP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3 și OCT2.
Cea mai mare parte a lenalidomidei este eliminată prin excreție urinară. Contribuția excreției renale la clearance-ul total la subiecții cu funcție renală normală a fost de 90%, în timp ce 4% din lenalidomidă a fost eliminată în materiile fecale.
Lenalidomida este slab metabolizată, astfel încât 82% din doză este excretată nemodificată în urină. Hidroxi-lenalidomida și N-acetil-lenalidomida reprezintă 4,59%, respectiv 1,83% din doza excretată. Clearance-ul renal al lenalidomidei depășește rata de filtrare glomerulară, prin urmare cel puțin într-o anumită măsură este secretată activ.
La doze de 5 până la 25 mg / zi, timpul de înjumătățire plasmatică este de aproximativ 3 ore la voluntarii sănătoși și variază de la 3 la 5 ore la pacienții cu mielom multiplu, sindroame mielodisplazice sau limfom cu celule de manta.
Pacienți vârstnici
Nu au fost efectuate studii clinice specifice pentru a evalua farmacocinetica lenalidomidei la pacienții vârstnici. Analizele farmacocinetice populaționale au inclus pacienți cu vârsta cuprinsă între 39 și 85 de ani și au indicat faptul că vârsta nu afectează clearance-ul (concentrația plasmatică) a lenalidomidei. Deoarece pacienții vârstnici sunt mai susceptibili de a avea funcție renală scăzută, se recomandă prudență în selectarea dozei și monitorizarea funcției renale este recomandată ca măsură de precauție.
Insuficiență renală
Farmacocinetica lenalidomidei a fost studiată la subiecții cu insuficiență renală cauzată de boli non-maligne. În acest studiu, s-au utilizat două metode pentru clasificarea funcției renale: clearance-ul creatininei urinare măsurat pe parcursul a 24 de ore și clearance-ul creatininei estimat cu formula Cockcroft-Gault. aproximativ 3,5 ore la subiecții cu clearance-ul creatininei> 50 ml / min până la mai mult de 9 ore la subiecții cu funcție renală afectată
Insuficiență hepatică
Analizele farmacocinetice ale populației au inclus pacienți cu insuficiență hepatică ușoară (N = 16, bilirubină totală> 1 până la ≤ 1,5 x ULN (Limita superioară a normalului) sau AST> ULN) și au indicat că „insuficiența hepatică ușoară nu afectează clearance-ul (concentrația plasmatică) de lenalidomidă Nu sunt disponibile date la pacienții cu insuficiență hepatică moderată până la severă.
Alți factori intrinseci
Analizele farmacocinetice populaționale indică faptul că greutatea corporală (33-135 kg), sexul, rasa și tipul de malignitate hematologică (MM, MDS sau MCL) nu au un efect relevant clinic asupra clearance-ului lenalidomidei la pacienții adulți.
05.3 Date preclinice de siguranță -
Un studiu de dezvoltare embrion-fetală a fost efectuat la maimuțele tratate cu lenalidomidă la doze cuprinse între 0,5 și 4 mg / kg / zi. Rezultatele acestui studiu au indicat faptul că lenalidomida provoacă malformații externe, inclusiv orificiu anal nebrevetat și malformații ale extremităților superioare și inferioare (părți ale extremităților curbate, scurtate, malformate, malrotate și / sau absente, oligo și / sau polidactilie) la descendenți.maimuțelor de sex feminin care au primit medicamentul în timpul gestației.
S-au observat, de asemenea, diferite efecte viscerale la fetuși individuali (decolorare, focare roșii în diferite organe, masă mică incoloră deasupra valvei atrioventriculare, vezică biliară mică, diafragmă malformată).
Lenalidomida prezintă un risc potențial de toxicitate acută; la rozătoare, dozele letale minime după administrarea orală au fost> 2.000 mg / kg / zi. Administrarea orală repetată de 75, 150 și 300 mg / kg / zi timp de până la 26 de săptămâni a dus la o creștere reversibilă a mineralizării pelvine renale legate de tratament la șobolani, în principal femele, la toate nivelurile de doză. Nivelul fără efecte adverse observabile (NOAEL, niciun nivel de efect advers observat) a fost considerat a fi mai mic de 75 mg / kg / zi și este de aproximativ 25 de ori mai mare decât expunerea zilnică la om pe baza valorilor ASC. La maimuțe, administrarea orală repetată de 4 și 6 mg / kg / zi pentru perioade de până la 20 de săptămâni a produs mortalitate și toxicitate semnificative (scădere semnificativă în greutate, număr redus de globule albe, număr de globule roșii și trombocite, hemoragii în mai multe organe, inflamație a tractul gastrointestinal, atrofia țesutului limfatic și a măduvei osoase). De asemenea, la maimuțe, administrarea orală repetată de 1 și 2 mg / kg / zi, pentru perioade de până la 1 an, a produs modificări reversibile ale celularității măduvei osoase, o ușoară reducere a raportului celulelor mielo-eritroide și atrofie timică. O scădere ușoară a numărului de celule albe din sânge a fost observată la 1 mg / kg / zi, ceea ce corespunde aproximativ aceleiași doze la om, pe baza comparației ASC.
Studii de mutagenitate efectuate in vitro (mutație bacteriană, limfocite umane, limfom murin, transformare în celule embrionare de hamster sirian) și in vivo (testul micronucleului de șobolan) nu a evidențiat niciun efect legat de medicament, nici la nivelul genei, nici la nivelul cromozomului. Nu s-au efectuat studii de cancerigenitate cu lenalidomidă.
Toxicitatea asupra dezvoltării a fost studiată anterior la iepuri. În aceste studii, iepurilor li s-au administrat oral 3, 10 și 20 mg / kg / zi de lenalidomidă. Absența lobului intermediar pulmonar a fost observată la doza de 10 și 20 mg / kg / zi, cu o corelație cu doza și rinichiul ectopic la doza de 20 mg / kg / zi. Deși aceste condiții au fost observate la o doză de 20 mg / kg / zi. doze toxice pentru mamă, acestea pot fi atribuite unui efect direct La doze de 10 și 20 mg / kg / zi, modificări ale țesuturilor moi și ale scheletului au fost observate și la făt.
06.0 INFORMAȚII FARMACEUTICE -
06.1 Excipienți -
Conținutul capsulei
Lactoză anhidră
Celuloză microcristalină
Croscarmeloză sodică
Stearat de magneziu
Coaja capsulei
Jeleu
Dioxid de titan (E171)
Carmin indigo (E132)
Oxid de fier galben (E172)
Cerneala formulării
Şerlac
Propilen glicol
Oxid de fier negru (E172)
Hidroxid de potasiu
06.2 Incompatibilitate "-
Nu este relevant.
06.3 Perioada de valabilitate "-
3 ani.
06.4 Precauții speciale pentru depozitare -
Acest medicament nu necesită condiții speciale de păstrare.
06.5 Natura ambalajului imediat și conținutul ambalajului -
Clorură de polivinil (PVC) / policlorotrifluoretilenă (PCTFE) / blister din folie de aluminiu conținând 7 capsule dure.
Pachet de 21 capsule.
06.6 Instrucțiuni de utilizare și manipulare -
Medicamentele neutilizate și deșeurile derivate din acest medicament trebuie eliminate în conformitate cu reglementările locale.
07.0 DEȚINĂTORUL „AUTORIZAȚIEI DE PUNERE PE PIAȚĂ” -
Celgene Europe Limited
1 drum lung
Stockley Park
Uxbridge
UB11 1DB
Regatul Unit
08.0 NUMĂRUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ -
EU / 1/07/391/002
038016022
09.0 DATA PRIMEI AUTORIZAȚII SAU REÎNNOIREA AUTORIZAȚIEI -
Data primei autorizații: 14 iunie 2007
Data celei mai recente reînnoiri: 14 iunie 2012
10.0 DATA REVIZUIRII TEXTULUI -
D.CCE septembrie 2016
11.0 PENTRU RADIOFarmaceutice, DATE COMPLETE PRIVIND DOSIMETRIA INTERNĂ DE RADIARE -
12.0 PENTRU DROGURILE RADIO, INSTRUCȚIUNI SUPLIMENTARE PRELUCRATE PREPARĂRI EXTEMPORALE ȘI CONTROLUL CALITĂȚII -





-cause-sintomi-e-terapia.jpg)