Generalitate
Sindromul Crouzon este o boală genetică rară, care determină prezența craniosinostozei și a altor anomalii faciale destul de deosebite.
Apariția sa este cauzată de anumite modificări ale ADN-ului care constituie genele FGFR2 și FGFR3; aceste elemente genetice sunt implicate în procesul de maturare a oaselor în timpul dezvoltării embrionare.

Terapia constă dintr-o serie de intervenții chirurgicale, menite să rezolve cele mai importante și mai periculoase simptome.
În prezent, prognosticul tinde să fie foarte des pozitiv.
Revizuirea geneticii
Înainte de a continua cu descrierea sindromului Crouzon, este util să se revizuiască câteva concepte fundamentale de genetică.
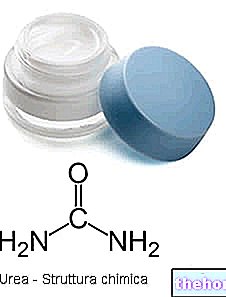
Ce este ADN-ul? Este patrimoniul genetic, în care sunt scrise trăsăturile somatice, predispozițiile, calitățile fizice, caracterul etc. al unui organism viu. Este conținut în toate celulele corpului care au un nucleu, ca se află în aceasta.
Ce sunt cromozomii? Conform definiției, cromozomii sunt unitățile structurale în care ADN-ul este organizat. Celulele umane conțin, în nucleul lor, 23 de perechi de cromozomi omologi (22 de tip autosomal non-sexual și o pereche de tip sexual); fiecare pereche este diferită de alta, deoarece conține o secvență genetică specifică.
Ce sunt genele? Acestea sunt scurte întinderi sau secvențe de ADN cu o semnificație biologică fundamentală: din ele, de fapt, derivă proteine sau molecule biologice fundamentale pentru viață. În gene, există o parte „scrisă” din ceea ce suntem și cine vom deveni.
Fiecare genă este prezentă în două versiuni, alelele: o alelă este de origine maternă, prin urmare transmisă de mamă; cealaltă alelă este de origine paternă, prin urmare transmisă de tată.
Ce este o mutație genetică? Este o greșeală în secvența ADN, care formează o genă. Din cauza acestei erori, proteina rezultată este fie defectă, fie complet absentă. În ambele cazuri, efectele pot fi dăunătoare atât pentru viața celulei, în care apare mutația, cât și pentru cea a organismului în ansamblu. Bolile congenitale și neoplasmele (adică tumorile) aparțin uneia sau mai multor mutații genetice.
La fel și sindromul Crouzon
Sindromul Crouzon este o afecțiune genetică rară caracterizată prin craniosinostoză și „dezvoltarea nefirească a anumitor elemente faciale, inclusiv ochii, nasul, maxilarul și maxilarul”.
Este o boală congenitală, ale cărei caracteristici tipice pot fi evidente deja în primele momente ale vieții.
SIGNIFICAT DE CRANIOSINOSTOSI
Craniosinostoză este termenul prin care medicii se referă la fuziunea prematură a uneia sau mai multor suturi craniene.

De pe site: thecraniofacialcenter.com
Suturile craniene sunt articulațiile fibroase care unesc oasele bolții craniene împreună (adică oasele frontale, temporale, parietale și occipitale).
În condiții normale, fuziunea suturilor craniene are loc în perioada post-natală (unele procese se încheie chiar la vârsta de 20 de ani). Acest lung proces de fuziune permite creierului să crească și să se dezvolte în mod adecvat.
Dacă, la fel ca în cazul craniosinostozelor, fuziunea are loc prea devreme - deci în timpul vieții prenatale, perinatale * sau a copilăriei timpurii - elementele creierului (creierul, cerebelul și trunchiul cerebral) și unele organe ale simțurilor (în special ochii) suferă o „alterare” de formă și creștere.
* Termenul perinatal se referă la perioada de viață care merge de la a 27-a săptămână de gestație până la primele 28 de zile după naștere.
ORIGINEA NUMEI
Sindromul Crouzon își datorează numele medicului francez Octave Crouzon, care are meritul de a fi descris mai întâi principalele sale caracteristici clinice.
Crouzon a trăit între sfârșitul anilor 1800 și începutul anilor 1900, exact din 1874 până în 1938. Inițial, pentru a defini sindromul care i-a luat ulterior numele, el a folosit termenul de disostoză craniofacială.
Cauze
Sindromul Crouzon apare ca urmare a unei mutații a genei FGFR2, localizată pe cromozomul 10 sau a genei FGFR3, localizată pe cromozomul 4.
FGFR este acronimul în engleză pentru Receptorul factorului de creștere a fibroblastelor, care s-a tradus în italiană este: Receptor pentru factorul de creștere a fibroblastelor.
Rolul funcțional al genelor FGFR2 și FGFR3 este fiecare de a produce o proteină receptoră, care, la rândul său, are sarcina de a regla maturarea și dezvoltarea embrionară a țesutului osos.
Potrivit teoriilor cercetătorilor, mutațiile din FGFR2 și FGFR3 ar hiper-stimula aceste aceleași gene, care, odată mai active, ar induce o maturare timpurie a unor țesuturi osoase, inclusiv a celor care constituie craniul.
GENETICA
Mutațiile genetice responsabile de sindromul Crouzon pot fi ereditare sau pot apărea spontan după concepție.
În primul caz, starea morbidă - la care medicii se referă și ca sindrom Crouzon ereditar - are toate caracteristicile unei boli genetice autosomale dominante (sau boală dominantă moștenită). Pentru un cititor începător de genetică, aceasta înseamnă că:
- Boala și simptomele acesteia apar și în prezența unei singure alele genetice mutante (nu contează dacă provine de la mamă sau tată), deoarece aceasta din urmă este dominantă asupra celei sănătoase.
- Un părinte care poartă mutația este suficient pentru a avea boala într-o parte din descendenți.
- Probabilitatea ca un copil bolnav să se nască, dintr-un cuplu în care doar una dintre cele două componente poartă mutația, este de 50%.
În cel de-al doilea caz, însă, starea morbidă - pe care experții o indică cu terminologia sindromului Crouzon non-ereditar - este rezultatul unui eveniment sporadic anomal, care modifică ADN-ul în timpul creșterii embrionare a fătului.
Rezumatul semnificației termenilor ereditari, autozomali și dominanți
- Ereditar: înseamnă că părinții transmit alterarea genetică responsabilă de boală descendenților (adică copiilor).
- Autosomal: înseamnă că mutația responsabilă de boală se află într-un cromozom non-sexual, deci autosomal.
- Dominant: înseamnă că boala provoacă simptome și semne chiar și atunci când o singură alelă a genei responsabile este mutată. În termeni mai simpli, este ca și cum alela cu mutația ar avea mai multă putere decât alela sănătoasă.
EPIDEMIOLOGIE
Conform unor estimări ale ratei de incidență a sindromului Crouzon, aproximativ unul din 60.000 de copii s-ar naște cu această afecțiune rară.
Sindromul Crouzon reprezintă 4,5% din cazurile de craniosinostoză.
Simptome și complicații
Pacienții cu sindrom Crouzon au o imagine a simptomelor foarte specifică, care constă de obicei din:
- Probleme legate de craniosinostoză, inclusiv:
-

De pe https://en.wikipedia.org/wiki/Plagiocephaly Brahicefalie, care este stoarcerea spatelui capului. Urmează fuziunea prematură a suturilor craniene coronare (craniosinostoză coronară).
Dacă nu este tratată, poate afecta creșterea creierului și dezvoltarea abilităților cognitive.
Ele reprezintă o „alternativă la brahicefalie: trigonocefalia (fuziunea suturii metopice), dolichocefalia (fuziunea suturii sagittale) și plagiocefalia (fuziunea suturilor coronare). - Exoftalmie, care este termenul pentru proeminența globilor oculari. Ar putea implica prezența problemelor de vedere.
- Hipertelorismul ocular, adică ochi care sunt exagerat de îndepărtați unul de celălalt. Cu exoftalmie, poate agrava problemele de vedere.
- Nas deformat, în general în formă de cioc. Dacă este severă sau nu este tratată chirurgical, această anomalie poate duce la probleme de respirație sau la aceleași simptome ca sindromul de apnee obstructivă în somn.
- Creșterea presiunii intracraniene. Este, de asemenea, cunoscut sub numele de hipertensiune intracraniană. Prezența sa se explică prin faptul că structurile creierului nu au spațiul potrivit pentru a crește.
De obicei întâlnită la mijlocul copilăriei târzii, hipertensiunea intracraniană este o cauză potențială de cefalee, vărsături și dureri oculare. - Hidrocefalie, care este rezultatul unei creșteri a lichidului cefalorahidian conținut în spațiul subarahnoidian și în ventriculii cerebrali.
- Malformația Arnold-Chiari (sau sindromul Arnold-Chiari). Este o deformare situată la baza craniului.
* Hidrocefalia și malformația Arnold-Chiari sunt, în general, două complicații care apar în absența tratamentelor adecvate. - Anomalii la nivelul mandibulei și maxilarului.
Primul are dimensiuni mai mici decât în mod normal, în timp ce al doilea tinde să iasă în afară. Toate acestea schimbă forma palatului și a schelei dentare (absența unor dinți etc.), cu repercusiuni (uneori chiar grave) asupra fonării și asupra mestecând.
Unii pacienți se nasc cu buza despicată (buza despicată) sau cu despicătura palatului.
- Probleme de auz.
55% dintre pacienții cu sindrom Crouzon se nasc fără canale urechii sau cu anomalii majore în acestea. Aceasta are ca rezultat o capacitate acustică absentă sau foarte redusă.
Unii subiecți dezvoltă un set de probleme de auz la vârsta adultă, atribuibile tabloului clinic tipic al sindromului Ménière.
- Probleme articulare la nivelul gâtului.
Acestea privesc 30% din cazurile de sindrom Crouzon.
- Anomalii cutanate.
Sunt prezenți pacienți cu sindrom Crouzon mutat cu FGFR3 acanthosis nigricans, o dermatoză caracterizată prin creșterea grosimii (hiperkeratoza) și întunecarea (hiperpigmentarea) pielii.

Alte două anomalii anatomice asociate (deși rar) cu sindromul Crouzon
- Canalul arterial brevetat
- Coarctația aortei
SINDROMUL CROUZON ȘI IQ
Datorită și posibilităților actuale de tratare a craniosinostozei, astăzi 97% dintre pacienții cu sindrom Crouzon au „inteligență normală”.
Diagnostic
Un medic pediatru cu experiență poate fi capabil să diagnosticheze sindromul Crouzon doar prin examinarea fizică.
În prezența oricărei îndoieli sau nedumeriri, următoarele sunt fundamentale pentru a ajunge la o concluzie precisă:
- Imagini radiologice, furnizate de raze X sau CT ale capului
- Un test genetic, menit să caute orice mutație a ADN-ului.
EXAMINARE OBIECTIVĂ
Examenul fizic constă într-o analiză exactă a capului și a anomaliilor prezente pe acesta.
Deformitățile craniene, induse de craniosinostoză (de exemplu brahicefalie), se numără printre cele mai caracteristice semne clinice ale sindromului Crouzon și pe care medicul își bazează o parte din concluziile sale diagnostice.
EXAMENE RADIOLOGICE
Razele X și CT ale capului arată care suturi craniene s-au contopit prematur.
Craniosinostozele care caracterizează sindromul Crouzon afectează suturile coronare, prin urmare o fuziune constatată la nivelul acestora din urmă este o informație foarte adesea decisivă în scopuri de diagnostic.
EXAMENUL GENETIC
Pe lângă faptul că arată dacă ADN-ul are mutații, testarea genetică ajută la identificarea genei exacte care cauzează sindromul Crouzon, indiferent dacă FGFR2 sau FGFR3.
Tratament
Astăzi, purtătorii sindromului Crouzon se pot baza pe diferite tratamente, în funcție de gravitatea afecțiunii și de simptome.
De fapt, medicii s-au asigurat:
- Chirurgie pentru rezolvarea craniosinostozei și a simptomelor acesteia.
- Ajutoare acustice, în caz de probleme de auz.
- Terapii pentru îmbunătățirea abilităților lingvistice.
- Terapii chirurgicale pentru ameliorarea anomaliilor la nivelul maxilarului și mandibulei.
- O intervenție chirurgicală, cunoscută sub numele de traheostomie, pentru rezolvarea problemelor de respirație.
Vă rugăm să rețineți: Sindromul Crouzon este o afecțiune morbidă care derivă dintr-o „alterare genetică a ADN-ului imposibil de vindecat. Deci, de fapt, medicii tratează boala doar din punct de vedere simptomatologic.
CHIRURGIE PENTRU CRANIOSINOSTOZĂ
Obiectivele terapeutice ale operației chirurgicale sunt două:
- Oferiți structurilor creierului și ochilor spațiul de care au nevoie pentru a se dezvolta și pentru a funcționa la maximum.
- Dați capului o formă normală, apoi rezolvați problema brahicefaliei.
Chirurgii au posibilitatea de a efectua operația în două moduri (sau abordări) diferite: printr-o „operație chirurgicală tradițională - numită și„ deschisă ”- sau printr-o„ operație chirurgicală endoscopică.
„Chirurgia deschisă” implică „executarea unei” incizii pe cap, prin care medicul operator operează extractul osului sau oaselor craniene malformate care trebuie remodelate. La sfârșitul remodelării, chirurgul reintroduce structurile osoase extrase anterior și închide incizia cu suturi.
Chirurgia endoscopică, pe de altă parte, implică utilizarea unui endoscop și practicarea unei incizii foarte mici pe cap, prin care medicul operator operează inserarea endoscopului în sine.
Endoscopul este de fapt un tub subțire și flexibil, echipat cu o cameră cu fibră optică (la capătul introdus în craniu) și conectat la un monitor. Prin acest instrument particular și imaginile pe care le proiectează pe monitor, chirurgul este capabil să separe suturile craniene care fuzionează prematur, cu o precizie remarcabilă și fără a recurge la incizii ale pielii și extracții osoase.
Potrivit experților, cel mai bun moment pentru a efectua intervenția chirurgicală este în copilăria foarte timpurie (primele 12 luni de viață), deoarece oasele se mulează mai ușor.
Cu toate acestea, trebuie amintit că cu cât pacientul este mai tânăr, cu atât este mai mare riscul unei recidive a acelorași suturi craniene (recurență) .În caz de recurență, intervenția chirurgicală trebuie repetată.
Conform unor cercetări statistice, 10-20% dintre subiecții foarte tineri, supuși unei intervenții chirurgicale pentru craniosinostoză, trebuie să fie supuși unei a doua operații, din cauza unei recăderi.
TRATAMENTUL PROBLEMELOR ACUSTICE
Pe lângă prescrierea utilizării aparatelor auditive, medicii recomandă și controale auditive periodice, deoarece acesta este cel mai bun mod de a preveni orice înrăutățire a problemelor existente.
TERAPII CHIRURGICALE PENTRU FALCĂ ȘI ANOMALII FALCĂ
Tratamentul anomaliilor maxilare și mandibulare include o intervenție chirurgicală pentru realinierea maxilarului și / sau a mandibulei, unele tratamente dentare pentru aranjarea arcadelor dentare și operația pentru rezoluția buzei fisurii și / sau a palatului fisurat.
TRAHEOSTOMIE
Traheostomia este operația chirurgicală prin care medicul creează, la nivelul gâtului (pe unde trece traheea), un pasaj pentru aerul destinat plămânilor. Acest lucru permite celor care sunt supuși acestei intervenții chirurgicale să respire din nou și corect.
Pentru a transporta aerul în plămâni, aveți nevoie de un tub mic, numit tub de transcheostomie, care are dimensiunea potrivită pentru introducerea în trahee.
Prognoză
În general, prognosticul depinde de severitatea craniosinostozei: dacă aceasta din urmă este tratabilă cu rezultate bune, pacienții cu sindrom Crouzon se pot bucura de o viață aproape normală.















.jpg)


-perch-e-quando-si-misura.jpg)