Ingrediente active: Infliximab
Remicade 100 mg pulbere pentru concentrat pentru soluție perfuzabilă
De ce se utilizează Remicade? Pentru ce este?
Remicade conține o substanță activă numită infliximab. Infliximab este o proteină de origine umană și animală (de la șoarece).
Remicade aparține unui grup de medicamente numite „blocante TNF”. Este utilizat la adulți pentru tratamentul următoarelor boli inflamatorii:
- Artrita reumatoida
- Artrita psoriazică
- Spondilita anchilozantă (boala Bechterew)
- Psoriazis.
Remicade este, de asemenea, utilizat la adulți și copii cu vârsta de 6 ani și peste pentru:
- Boala Crohn
- Colită ulcerativă.
Remicade funcționează prin blocarea acțiunii unei proteine numite „factor de necroză tumorală alfa” (TNFα). Această proteină este implicată în procesele inflamatorii ale corpului și prin blocarea acesteia, este posibilă reducerea inflamației în organism.
Artrita reumatoida
Artrita reumatoidă este o boală inflamatorie a articulațiilor. Dacă aveți artrită reumatoidă, veți fi tratat inițial cu alte medicamente. Dacă nu răspundeți în mod adecvat la aceste medicamente, veți fi tratat cu Remicade în asociere cu un alt medicament numit metotrexat pentru:
- Reduceți semnele și simptomele bolii,
- Încetiniți progresia deteriorării articulațiilor,
- Îmbunătățiți funcția fizică.
Artrita psoriazică
Artrita psoriazică este o boală inflamatorie a articulațiilor, de obicei însoțită de psoriazis. Dacă aveți artrită psoriazică, veți fi tratat mai întâi cu alte medicamente. Dacă nu răspundeți în mod adecvat la aceste medicamente, veți fi tratat cu Remicade pentru:
- Reduceți semnele și simptomele bolii,
- Încetiniți progresia deteriorării articulațiilor,
- Îmbunătățiți funcția fizică.
Spondilita anchilozantă (boala Bechterew)
Spondilita anchilozantă este o boală inflamatorie a coloanei vertebrale. Dacă aveți spondilită anchilozantă, veți fi tratat mai întâi cu alte medicamente. Dacă nu răspundeți în mod adecvat la aceste medicamente, veți fi tratat cu Remicade pentru:
- Reduceți semnele și simptomele bolii,
- Îmbunătățiți funcția fizică.
Psoriazis
Psoriazisul este o boală inflamatorie a pielii. Dacă aveți psoriazis în plăci moderat până la sever, veți fi tratat mai întâi cu alte medicamente sau alte tratamente, cum ar fi fototerapia. Dacă nu răspundeți în mod adecvat la aceste medicamente sau tratamente, veți fi tratat cu Remicade pentru a reduce semnele și simptomele bolii dumneavoastră.
Colită ulcerativă
Colita ulcerativă este o boală inflamatorie a intestinului. Dacă aveți colită ulcerativă, veți fi tratat mai întâi cu alte medicamente. Dacă nu răspundeți în mod adecvat la aceste medicamente, vi se va administra Remicade pentru tratarea bolii.
Boala Crohn
Boala Crohn este o boală inflamatorie a intestinului. Dacă aveți boala Crohn, veți fi tratați mai întâi cu alte medicamente. Dacă nu răspundeți în mod adecvat la aceste medicamente, veți fi tratat cu Remicade pentru: • Tratarea bolii Crohn active • Reducerea numărului de deschideri anormale (fistule) între intestin și piele, pentru care alte medicamente sau intervenții chirurgicale s-au dovedit inadecvate.
Contraindicații atunci când Remicade nu trebuie utilizat
Nu vi se va administra Remicade dacă:
- sunteți alergic la infliximab (substanță activă din Remicade) sau la oricare dintre celelalte componente ale acestui medicament (enumerate la punctul 6)
- sunteți alergic (hipersensibil) la proteinele de șoarece
- aveți tuberculoză (TB) sau o „altă infecție gravă, cum ar fi pneumonie sau sepsis
- aveți „insuficiență cardiacă moderată sau severă.
Nu luați Remicade dacă vi se aplică oricare dintre condițiile de mai sus. Dacă nu sunteți sigur, discutați cu medicul dumneavoastră înainte de a vi se administra Remicade
Precauții pentru utilizare Ce trebuie să știți înainte de a lua Remicade
Discutați cu medicul dumneavoastră înainte de a vi se administra Remicade dacă aveți:
A primit anterior Remicade
- Spuneți medicului dumneavoastră dacă ați avut tratament cu Remicade în trecut și dacă reluați tratamentul cu Remicade.
Dacă ați încetat să mai luați Remicade mai mult de 16 săptămâni, există un risc crescut de reacții alergice atunci când reporniți Remicade.
Infecții
Spuneți medicului dumneavoastră dacă aveți o „infecție, chiar și foarte minoră, înainte de a vi se administra Remicade
- Spuneți medicului dumneavoastră înainte de a vi se administra Remicade dacă ați locuit sau ați călătorit într-o „zonă în care infecțiile numite histoplasmoză, coccidioidomicoză sau blastomicoză sunt frecvente. Aceste infecții sunt cauzate de tipuri specifice de ciuperci care pot afecta plămânii sau alte părți ale corp.corp
- Puteți fi mai predispus la infecții atunci când sunteți tratat cu Remicade. Dacă aveți 65 de ani sau mai mult, aveți un risc mai mare
- Aceste infecții pot fi grave și includ tuberculoză, infecții cauzate de viruși, ciuperci sau bacterii sau alte infecții oportuniste și sepsis care, în cazuri rare, pot pune viața în pericol.
Spuneți imediat medicului dumneavoastră dacă aveți simptome de infecție în timpul tratamentului cu Remicade. Simptomele includ febră, tuse, simptome asemănătoare gripei, senzație de rău, piele roșie sau foarte fierbinte, răni sau probleme dentare. Medicul dumneavoastră vă poate recomanda oprirea temporară a Remicade.
Tuberculoza (TB)
- Este foarte important să spuneți medicului dumneavoastră dacă ați avut vreodată tuberculoză sau dacă ați fost în contact strâns cu persoane care au avut sau au tuberculoză
- Medicul dumneavoastră vă va face teste pentru a vedea dacă aveți tuberculoză. Câteva cazuri de tuberculoză au fost raportate la pacienții tratați cu Remicade, în rare ocazii chiar și la pacienții care au fost tratați cu medicamente pentru TBC. Medicul va înregistra aceste teste pe cardul de alertă al pacientului
- Dacă medicul dumneavoastră crede că sunteți expus riscului de tuberculoză, este posibil să fiți tratat cu medicamente pentru tuberculoză înainte de a vi se administra Remicade.
Spuneți imediat medicului dumneavoastră dacă observați semne de tuberculoză în timp ce luați Remicade. Semnele includ tuse persistentă, scădere în greutate, senzație de oboseală, febră, transpirații nocturne.
Virusul hepatitei B (VHB)
- Spuneți medicului dumneavoastră dacă sunteți purtător sau aveți sau ați avut hepatită B înainte de a vi se administra Remicade
- Spuneți medicului dumneavoastră dacă credeți că ați putea fi expus riscului de a contracta hepatita B
- Medicul trebuie să evalueze dacă aveți hepatită B? Tratamentul cu blocante TNF, cum ar fi Remicade, poate determina reactivarea virusului hepatitei B la pacienții cu acest virus, care, în unele cazuri, poate provoca moartea.
Probleme cu inima
- Spuneți medicului dumneavoastră dacă aveți probleme cardiace, cum ar fi insuficiență cardiacă ușoară
- Medicul dumneavoastră vă va monitoriza îndeaproape funcția inimii.
Spuneți imediat medicului dumneavoastră dacă observați semne noi sau agravante ale insuficienței cardiace în timpul tratamentului cu Remicade. Semnele includ dificultăți de respirație sau umflarea picioarelor.
Cancer și limfom
- Spuneți medicului dumneavoastră dacă aveți sau ați avut vreodată limfom (un tip de cancer de sânge) sau alte tipuri de cancer înainte de a vi se administra Remicade
- Pacienții cu poliartrită reumatoidă severă care au suferit de această boală pentru o lungă perioadă de timp pot avea un risc mai mare decât media de a dezvolta limfom.
- Copiii și adulții care iau Remicade pot prezenta un risc crescut de a dezvolta limfom sau un alt tip de cancer.
- Unii pacienți care au fost tratați cu blocante TNF, inclusiv Remicade, au dezvoltat un tip rar de cancer numit limfom cu celule T. Hepatosplenic. Majoritatea acestor pacienți au fost adolescenți sau bărbați tineri și majoritatea au avut colită Crohn sau ulcerativă. Acest tip de cancer este de obicei letal. Aproape toți pacienții au fost tratați și cu medicamente numite azatioprină sau 6-mercaptopurină, în plus față de blocantele TNF.
- Unii pacienți tratați cu infliximab au dezvoltat anumite tipuri de cancer de piele. Dacă aveți orice fel de schimbare a aspectului pielii sau creșteri pe piele în timpul sau după tratament, vă rugăm să spuneți medicului dumneavoastră.
Boli pulmonare sau fumat intens
- Spuneți medicului dumneavoastră dacă aveți o boală pulmonară numită boală pulmonară obstructivă cronică (BPOC) sau dacă sunteți un fumător intens înainte de a vi se administra Remicade
- Pacienții cu BPOC și care fumează intens pot prezenta un risc crescut de cancer atunci când sunt tratați cu Remicade.
Boala sistemului nervos
- Spuneți medicului dumneavoastră dacă aveți sau ați avut vreodată o problemă a sistemului nervos înainte de a vi se administra Remicade. Aceasta include scleroza multiplă, sindromul Guillain-Barré, atacurile sau diagnosticul de „nevrită optică”.
Spuneți imediat medicului dumneavoastră dacă observați simptome ale unei boli nervoase în timp ce luați Remicade. Semnele includ modificări ale vederii, slăbiciune în brațe și picioare, amorțeală sau furnicături în orice parte a corpului.
Deschideri anormale ale pielii
- Spuneți medicului dumneavoastră dacă aveți orificii anormale ale pielii (fistule) înainte de a vi se administra Remicade.
Vaccinări
- Spuneți medicului dumneavoastră dacă ați fost vaccinat recent sau intenționați să fiți vaccinat
- Nu trebuie să primiți niciun vaccin în timpul tratamentului cu Remicade
- Unele vaccinări pot provoca infecții. Dacă ați primit Remicade în timp ce ați fost însărcinată, bebelușul dumneavoastră poate avea un risc crescut de a face această infecție timp de aproximativ șase luni de la ultima doză primită în timpul sarcinii. când copilul dumneavoastră ar trebui să primească vaccinuri.
Agenți terapeutici infecțioși
- Discutați cu medicul dumneavoastră dacă ați luat recent sau intenționați să luați tratament cu un agent terapeutic infecțios (cum ar fi instilația BCG utilizată pentru tratarea cancerului).
Operații sau proceduri dentare
- Spuneți medicului dumneavoastră dacă urmează să aveți proceduri sau tratamente dentare
- Spuneți chirurgului sau dentistului care efectuează procedura că sunteți tratat cu Remicade prezentând cardul de alertă pentru pacient.
Copii și adolescenți
Informațiile de mai sus se aplică și copiilor și adolescenților. În plus:
- unii copii și adolescenți care au luat medicamente care blochează TNF, cum ar fi Remicade, au dezvoltat cancere, inclusiv tipuri neobișnuite, care au fost uneori letale.
- În comparație cu adulții, mai mulți copii care iau Remicade au dezvoltat infecții
- Copiii trebuie să primească vaccinările recomandate înainte de a începe tratamentul cu Remicade.
Dacă nu sunteți sigur dacă oricare dintre condițiile de mai sus vi se aplică, contactați medicul înainte de a vi se administra Remicade.
Interacțiuni Ce medicamente sau alimente pot schimba efectul Remicade
Pacienții cu boli inflamatorii iau deja medicamente pentru tratarea bolii. Aceste medicamente pot provoca reacții adverse. Medicul dumneavoastră vă va sfătui ce alte medicamente trebuie să continuați să luați în timp ce sunteți tratat cu Remicade.
Spuneți medicului dumneavoastră dacă luați sau ați luat recent orice alte medicamente, inclusiv orice alte medicamente pentru tratamentul bolii Crohn, colitei ulcerative, poliartritei reumatoide, spondilitei anchilozante, artritei psoriazice sau psoriazisului sau medicamentelor pe care le primiți fără prescripție medicală, precum vitamine și plante medicamente.
Spuneți în special medicului dumneavoastră dacă utilizați oricare dintre aceste medicamente:
- Medicamente care afectează sistemul imunitar
- Kineret (anakinra). Remicade și Kineret nu trebuie administrate împreună
- Orencia (abatacept). Remicade și Orencia nu trebuie administrate împreună.
Dacă nu sunteți sigur dacă oricare dintre condițiile de mai sus vi se aplică, contactați medicul înainte de a vi se administra Remicade.
Avertismente Este important să știm că:
Sarcina, alăptarea și fertilitatea
- Dacă sunteți gravidă sau alăptați, credeți că ați putea fi gravidă sau intenționați să rămâneți gravidă, adresați-vă medicului dumneavoastră pentru recomandări înainte de a lua acest medicament. Remicade nu este recomandat în timpul sarcinii
- Trebuie să evitați să rămâneți gravidă în timpul tratamentului cu Remicade și timp de cel puțin 6 luni după oprirea tratamentului.Asigurați-vă că utilizați contracepție adecvată în acest timp.
- Nu alăptați în timpul tratamentului cu Remicade sau timp de 6 luni după ultimul tratament cu Remicade
- Dacă ați primit Remicade în timpul sarcinii, bebelușul dumneavoastră poate avea un risc crescut de a face o infecție. Este important să spuneți medicului pediatru și altor profesioniști din domeniul sănătății despre utilizarea Remicade înainte ca bebelușul dumneavoastră să primească vaccinuri (pentru mai multe informații, consultați secțiunea privind vaccinările ).
Conducerea vehiculelor și utilizarea utilajelor
Este puțin probabil ca Remicade să vă afecteze capacitatea de a conduce vehicule sau de a folosi utilaje. Dacă vă simțiți obosit sau rău după tratamentul cu Remicade, nu trebuie să conduceți vehicule și să nu folosiți niciun instrument sau utilaje.
Doza, metoda și timpul de administrare Cum se utilizează Remicade: Doze
Cum se administrează Remicade
- Remicade vă va fi administrat de către medicul sau asistenta dumneavoastră
- Medicul sau asistenta dumneavoastră vă va pregăti soluția injectabilă Remicade
- Soluția Remicade va fi injectată încet (pe o perioadă de 2 ore) într-o venă, de obicei în braț. Această procedură se numește „perfuzie intravenoasă” sau picurare. După al treilea tratament, medicul dumneavoastră poate decide să vă administreze Remicade pe o perioadă de 1 oră
- Veți fi monitorizat în timpul administrării Remicade și timp de 1-2 ore după aceea.
Cât de mult este dat Remicade
- Medicul dumneavoastră va stabili doza (în mg) și intervalul dintre dozele de Remicade, care va depinde de boala, greutatea și răspunsul la tratament.
- Tabelul de mai jos arată frecvența administrării acestui medicament.
Artrita reumatoida
Doza uzuală este de 3 mg pentru fiecare kg de greutate corporală
Artrita psoriazică, spondilita anchilozantă (boala Bechterew), psoriazisul, colita ulcerativă și boala Crohn
Doza uzuală este de 5 mg pentru fiecare kg de greutate corporală.
Utilizare la copii și adolescenți
Remicade trebuie utilizat numai la copii pentru boala Crohn sau colita ulcerativă. Acești copii trebuie să aibă vârsta de 6 ani sau mai mult.
Supradozaj Ce trebuie făcut dacă ați luat prea mult Remicade
Dacă vi se administrează mai mult Remicade decât aveți nevoie
Deoarece acest medicament vi se administrează de către medicul sau asistenta medicală, este puțin probabil să primiți prea mult. Nu există efecte secundare cunoscute ale supradozajului Remicade.
Dacă uitați sau pierdeți o infuzie „Remicade”
Dacă uitați sau ratați o întâlnire pentru administrarea Remicade, faceți o altă întâlnire cât mai curând posibil.
Dacă aveți orice întrebări suplimentare cu privire la acest medicament, adresați-vă medicului dumneavoastră
Efecte secundare Care sunt efectele secundare ale Remicade
Ca toate medicamentele, acest medicament poate provoca reacții adverse, deși nu apar la toate persoanele. Majoritatea acestor efecte sunt ușoare până la moderate. Cu toate acestea, unii pacienți pot prezenta reacții adverse severe și necesită tratament medical. Efectele secundare pot apărea și după terminarea tratamentului cu Remicade.
Spuneți imediat medicului dumneavoastră dacă observați oricare dintre următoarele reacții adverse:
- Semne ale unei reacții alergice, cum ar fi umflarea feței, buzelor, gurii sau gâtului, care poate provoca dificultăți la înghițire sau respirație, erupții cutanate, urticarie, umflarea mâinilor, picioarelor sau gleznelor. Reacția alergică poate apărea în decurs de 2 ore de la injecție sau mai târziu. Alte semne ale unei reacții alergice care pot apărea până la 12 zile după injecție includ dureri musculare, febră, dureri articulare sau maxilare, dureri în gât sau dureri în gât.
- Semne ale unei probleme cardiace, cum ar fi dificultăți de respirație, umflarea picioarelor sau modificări ale bătăilor inimii
- Semne ale unei infecții (inclusiv tuberculoză), cum ar fi febră, senzație de oboseală, tuse (persistentă), respirație scurtă, simptome asemănătoare gripei, scădere în greutate, transpirații nocturne, diaree, răni, probleme dentare sau arsuri în timpul urinării
- Semne ale unei probleme pulmonare, cum ar fi tuse, dificultăți de respirație sau opresiune în piept
- Semne de probleme neurologice (inclusiv probleme oculare), cum sunt convulsii, furnicături sau amorțeli în orice parte a corpului, slăbiciune în brațe sau picioare, modificări ale vederii, cum ar fi vederea dublă sau alte probleme oculare
- Semne ale unei probleme hepatice, cum ar fi îngălbenirea pielii sau a ochilor, urină maro închis sau durere în partea dreaptă superioară a stomacului, febră
- Semne ale unei tulburări a sistemului imunitar, numită lupus, cum ar fi dureri articulare sau erupții cutanate pe obraji sau brațe, zone sensibile la soare
- Semne ale scăderii numărului de celule sanguine, cum ar fi febră persistentă, sângerări sau echimoze mai frecvente sau arătând palide.
Dacă observați oricare dintre simptomele descrise mai sus, spuneți imediat medicului dumneavoastră.
Reacții adverse foarte frecvente (afectează mai mult de 1 din 10 pacienți)
- Dureri de stomac, stare de rău
- Infecții virale, cum ar fi herpesul sau gripa
- Infecții ale tractului respirator superior, cum ar fi sinuzita
- Durere de cap
- Efect nedorit din cauza perfuziei
- Durere.
Reacții adverse frecvente (afectează 1 până la 10 utilizatori din 100)
- Modificări ale funcției hepatice, creșterea enzimelor hepatice (observate la testele de sânge)
- Infecții ale plămânilor sau ale pieptului, cum ar fi bronșita sau pneumonia
- Dificultăți de respirație sau durere la respirație, durere în piept
- Sângerări în stomac sau intestine, diaree, indigestie, arsuri la stomac, constipație
- Erupție asemănătoare urticariei, erupții cutanate cu mâncărime sau piele uscată
- Probleme cu echilibrul sau senzația de amețeală
- Febra, transpirație crescută
- Probleme de circulație, cum ar fi tensiunea arterială scăzută sau crescută
- Vânătăi, înroșire sau sângerări nazale, piele fierbinte, roșie (roșeață)
- Senzație de oboseală sau slăbiciune
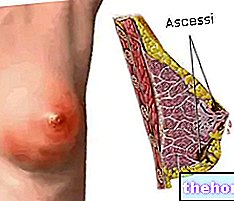
- Infecții bacteriene, cum ar fi infecția generalizată, abcesul sau infecția straturilor profunde ale pielii (celulită)
- Probleme de sânge, cum ar fi anemie sau număr scăzut de celule albe din sânge
- Ganglioni limfatici măriți
- Depresie, tulburări de somn
- Probleme oculare, inclusiv ochi roșii și infecții
- Bătăi rapide ale inimii (tahicardie) sau palpitații
- Durere la nivelul articulațiilor, mușchilor sau spatelui
- Infecții ale tractului urinar
- Psoriazis, probleme ale pielii, cum ar fi eczeme și căderea părului
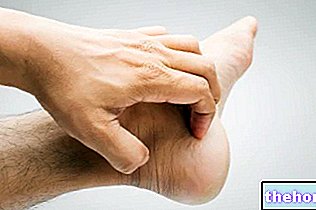
- Reacții la locul injectării, cum ar fi durere, umflături, roșeață sau mâncărime
- Frisoane, acumularea de lichid sub piele cauzând umflături
- Amorțeală sau senzație de furnicături.
Reacții adverse mai puțin frecvente (afectează 1 până la 10 utilizatori din 1000)
- Alimentare slabă de sânge, umflarea unei vene
- Probleme ale pielii, cum ar fi vezicule, veruci, decolorare anormală sau pigmentare a pielii sau buze umflate
- Reacții alergice severe (de exemplu, anafilaxie), tulburări ale sistemului imunitar numite lupus, reacții alergice la proteine străine
- Rani care se vindecă lent
- Umflarea ficatului (hepatită) sau a vezicii biliare (vezica biliară), afectarea ficatului
- Distragere, iritabilitate, confuzie, nervozitate
- Probleme oculare, incluzând vederea încețoșată sau redusă, umflarea ochilor sau stanții
- Insuficiență cardiacă nouă sau agravată, ritm cardiac lent
- Leșin
- Convulsii, tulburări nervoase
- Perforarea intestinului sau blocajul intestinal, dureri de stomac sau crampe
- Umflarea pancreasului (pancreatită)
- Infecții fungice, cum ar fi infecția cu drojdie
- Probleme pulmonare (cum ar fi edem)
- Lichid excesiv în jurul plămânilor (revărsat pleural)
- Infecții renale
- Număr scăzut de trombocite, număr excesiv de celule albe din sânge
- Infecții în vagin.
Reacții adverse rare (afectează 1 până la 10 utilizatori din 10.000)
- Un tip de cancer de sânge (limfom)
- Alimentare slabă de oxigen către organe prin sânge, probleme de circulație, cum ar fi îngustarea unui vas de sânge
- Inflamația membranei care acoperă creierul (meningită)
- Infecții datorate unui sistem imunitar slăbit
- Infecție cu hepatită B, dacă ați avut hepatită B în trecut? Umflarea sau creșterea țesuturilor anormale
- Umflarea vaselor de sânge mici (vasculită)? Tulburări imunologice care pot afecta plămânii, pielea și ganglionii limfatici (cum ar fi sarcoidoza)
- Lipsa de interes sau emoție
- Probleme grave ale pielii, cum ar fi necroliza epidermică toxică, sindromul Steven-Johnson sau eritemul multiform, probleme ale pielii, cum ar fi furunculele
- Tulburări grave ale sistemului nervos, cum ar fi mielita transversă, boala asemănătoare sclerozei multiple, nevrita optică și sindromul Guillain-Barré
- Fluid în membrana care acoperă inima (revărsat pericardic)
- Probleme pulmonare severe (cum ar fi pneumonia interstițială)
- Melanom (un tip de cancer de piele).
Alte reacții adverse (frecvența este necunoscută)
- Cancer la copii și adulți
- Un cancer de sânge rar care afectează în principal tinerii (limfom cu celule T hepatosplenice)
- Insuficiență hepatică
- Carcinom cu celule Merkel (un tip de cancer de piele)
- Agravarea unei afecțiuni numită dermatomiozită (arată ca o „erupție care însoțește slăbiciunea musculară).
Reacții adverse suplimentare la copii și adolescenți
Copiii care au luat Remicade pentru boala Crohn au prezentat unele diferențe în ceea ce privește efectele secundare comparativ cu adulții care au luat Remicade pentru boala Crohn.
Cele mai frecvente efecte secundare la copii au fost: număr scăzut de celule roșii din sânge (anemie), sânge în scaun, număr scăzut de celule albe din sânge (leucopenie), înroșire sau roșeață (bufeuri), infecții virale, număr scăzut de neutrofile (neutropenie) care sunt celule albe din sânge care combat infecțiile, fracturile osoase, infecțiile bacteriene și reacțiile alergice ale tractului respirator.
Raportarea efectelor secundare
Dacă manifestați orice reacții adverse, discutați cu medicul dumneavoastră, farmacistul sau asistenta medicală.Acestea includ orice posibile reacții adverse nemenționate în acest prospect. De asemenea, puteți raporta reacțiile adverse direct prin intermediul sistemului național de raportare enumerat în Anexa V. Reacțiile adverse pe care le puteți ajuta furnizați mai multe informații despre siguranța acestui medicament.
Expirare și reținere
Remicade va fi în general stocat de către profesioniștii din domeniul sănătății. Dacă aveți nevoie, detaliile de păstrare sunt următoarele:
- Nu lăsați acest medicament la vederea și îndemâna copiilor.
- Nu utilizați acest medicament după data de expirare înscrisă pe etichetă și cutie după „EXP”. Data de expirare se referă la ultima zi a lunii respective.
- A se păstra la frigider (2 ° C - 8 ° C).
- Acest medicament poate fi, de asemenea, păstrat în cutia originală în afara frigiderului până la maximum 25 ° C pentru o singură perioadă de până la șase luni. În această situație, nu trebuie păstrat din nou în frigider. Scrieți noua dată de expirare în casetă, inclusiv ziua / luna / anul. Aruncați acest medicament dacă nu este utilizat până la noua dată de expirare sau până la data de expirare tipărită pe cutie, oricare ar fi prima.
- Când Remicade este pregătit pentru perfuzie, se recomandă utilizarea acestuia cât mai curând posibil (în 3 ore). Cu toate acestea, dacă soluția este preparată în condiții complet fără germeni, poate fi păstrată la frigider timp de 24 de ore între 2 ° C și 8 ° C.
- Nu utilizați acest medicament dacă este decolorat sau are particule.
Ce conține Remicade
- Ingredientul activ este infliximab. Fiecare flacon conține 100 mg infliximab. După preparare, fiecare ml conține 10 mg infliximab.
- Celelalte componente sunt zaharoză, polisorbat 80, fosfat de sodiu monobazic și fosfat de sodiu dibazic.
Cum arată Remicade și conținutul ambalajului
Remicade este furnizat într-un flacon de sticlă care conține pulberea pentru concentrat pentru soluție perfuzabilă. Pulberea constă din granule albe liofilizate.
Remicade este disponibil în pachete de 1, 2, 3, 4 sau 5 flacoane. Nu toate mărimile de ambalaj pot fi comercializate
Prospect sursă: AIFA (Agenția italiană pentru medicamente). Conținut publicat în ianuarie 2016. Este posibil ca informațiile prezente să nu fie actualizate.
Pentru a avea acces la cea mai actualizată versiune, este recomandabil să accesați site-ul web AIFA (Agenția italiană pentru medicamente). Declinare de responsabilitate și informații utile.
01.0 DENUMIREA PRODUSULUI MEDICAMENTAL
REMICADE 100 mg pulbere pentru concentrat pentru soluție pentru perfuzie
02.0 COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
Fiecare flacon conține 100 mg infliximab. Infliximab este un anticorp monoclonal IgG1 uman-murin chimeric produs în celule de hibridom murin prin tehnologia ADN-ului recombinant. După reconstituire, fiecare ml conține 10 mg infliximab.
Pentru lista completă a excipienților, vezi secțiunea 6.1.
03.0 FORMA FARMACEUTICĂ
Pulbere pentru concentrat pentru soluție perfuzabilă.
Pulberea constă din granule albe liofilizate.
04.0 INFORMAȚII CLINICE
04.1 Indicații terapeutice
Artrita reumatoida
Remicade, în combinație cu metotrexat, este indicat pentru reducerea semnelor și simptomelor și îmbunătățirea funcției fizice în:
• pacienți adulți cu boală activă atunci când răspunsul la medicamentele antireumatice modificatoare de boală (DMARD), inclusiv metotrexatul, a fost inadecvat.
• pacienți adulți cu boală severă, activă și progresivă care nu au fost tratați anterior cu metotrexat sau alte DMARD.
O reducere a ratei de progresie a leziunilor articulare a fost demonstrată prin evaluarea radiografică la această populație de pacienți (vezi pct. 5.1).
Boala Crohn la adulți
Remicade este indicat pentru:
• tratamentul bolii Crohn active moderate până la severe la pacienții adulți care nu au reușit să răspundă în ciuda tratamentului complet și adecvat cu corticosteroizi și / sau imunosupresoare; sau la pacienții care nu tolerează sau au contraindicații medicale pentru terapiile menționate anterior.
• tratamentul bolii Crohn fistulizante active la pacienții adulți care nu au reușit să răspundă în ciuda unui curs complet și adecvat de terapie cu tratament convențional (inclusiv antibiotice, drenaj și terapie imunosupresoare).
Boala Crohn la copii
Remicade este indicat pentru tratamentul bolii Crohn active severe, la copii și adolescenți cu vârsta cuprinsă între 6-17 ani care nu au răspuns la terapia convențională cu un corticosteroid, un imunomodulator și o terapie nutrițională primară, sau la pacienții care nu tolerează sau au contraindicații pentru terapiile menționate mai sus. Remicade a fost studiat doar în combinație cu terapia convențională imunosupresoare.
Colită ulcerativă
Remicade este indicat pentru tratamentul colitei ulcerative active moderate până la severe la pacienții adulți care nu au răspuns adecvat la terapia convențională, inclusiv corticosteroizi și 6-mercaptopurină (6-MP) sau azatioprină (AZA), sau care sunt intoleranți sau pentru care există o contraindicație medicală a acestor terapii.
Colita ulcerativă la copii
Remicade este indicat pentru tratamentul colitei ulcerative active severe la copii și adolescenți cu vârsta cuprinsă între 6 și 17 ani care nu au răspuns adecvat la terapia convențională, inclusiv corticosteroizi și 6-MP sau AZA, sau care au intoleranță sau pentru care există un medic contraindicație la aceste terapii.
Spondilită anchilozantă
Remicade este indicat pentru tratamentul spondilita anchilozantă activă severă la pacienții adulți care nu au răspuns adecvat la terapiile convenționale.
Artrita psoriazică
Remicade este indicat pentru tratamentul artritei psoriazice active și progresive la pacienții adulți atunci când răspunsul la tratamentele DMARD anterioare a fost inadecvat.
Remicada trebuie administrată:
• în asociere cu metotrexat
• sau individual la pacienții care sunt intoleranți la metotrexat sau pentru care este contraindicat
S-a demonstrat că Remicade îmbunătățește funcția fizică la pacienții cu artrită psoriazică și reduce rata de progresie a leziunilor articulare periferice, măsurată prin raze X la pacienții cu subtipuri poliarticulare simetrice ale bolii (vezi pct. 5.1).
Psoriazis
Remicade este indicat pentru tratamentul psoriazisului plăcii moderat până la sever la pacienții adulți care au eșuat sau sunt contraindicați sau care au fost intoleranți la alte tratamente sistemice, inclusiv ciclosporină, metotrexat sau PUVA (vezi pct. 5.1).
04.2 Doze și mod de administrare
Tratamentul cu Remicade trebuie inițiat și supravegheat de către medici specialiști cu experiență în diagnosticul și tratamentul artritei reumatoide, a bolii inflamatorii intestinale, a spondilita anchilozantă, a artritei psoriazice sau a psoriazisului. Remicade trebuie administrat intravenos. Perfuziile cu Remicade trebuie administrate de personal medical calificat, instruit în recunoașterea oricăror probleme legate de perfuzie Pacienții tratați cu Remicade trebuie să primească prospectul și cardul de alertă al pacientului.
În timpul tratamentului cu Remicade, utilizarea altor terapii concomitente, cum ar fi corticosteroizii și imunosupresoarele, trebuie optimizată.
Dozare
Adulți (≥ 18 ani)
Artrita reumatoida
O perfuzie intravenoasă de 3 mg / kg urmată de perfuzii suplimentare de 3 mg / kg la săptămânile 2 și 6 după prima perfuzie, apoi la fiecare 8 săptămâni.
Remicade trebuie administrat concomitent cu metotrexat.
Datele disponibile sugerează că răspunsul clinic se obține de obicei în decurs de 12 săptămâni de la inițierea tratamentului. Dacă un pacient are un răspuns inadecvat sau pierde răspunsul după această perioadă, poate fi luată în considerare o creștere treptată a dozei de 1,5 mg / kg, până la un maxim de 7,5 mg / kg, la fiecare 8 săptămâni. Alternativ, ar putea fi luată în considerare administrarea a 3 mg / kg la fiecare 4 săptămâni. Dacă se obține un răspuns adecvat, tratamentul ar trebui să continue. Pacienții cu doza sau frecvența aleasă. continuați terapia la pacienții care nu prezintă dovezi de beneficii terapeutice în primele 12 săptămâni de tratament sau după ajustarea dozei.
Boala Crohn activă moderată până la severă
5 mg / kg administrat sub formă de perfuzie intravenoasă, urmat de o perfuzie suplimentară de 5 mg / kg la 2 săptămâni după prima perfuzie. Dacă un pacient nu răspunde la terapie după 2 doze, nu trebuie administrat un tratament suplimentar cu infliximab. Datele disponibile nu acceptă tratamentul suplimentar cu infliximab la non-pacienți respondenți în decurs de 6 săptămâni de la prima perfuzie.
La pacienții care răspund, soluțiile alternative pentru continuarea tratamentului sunt:
• Întreținere: perfuzie suplimentară de 5 mg / kg în săptămâna 6 după prima doză, urmată de perfuzii repetate la fiecare 8 săptămâni sau
• Re-administrare: o perfuzie de 5 mg / kg dacă persistă semnele și simptomele bolii (vezi pct. „Re-administrare” și pct. 4.4).
Deși lipsesc datele comparative, datele limitate la pacienții care au răspuns inițial la terapia cu 5 mg / kg, dar au pierdut răspunsul indică faptul că unii pacienți pot recupera răspunsul prin creșterea dozei (vezi pct. 5.1). Tratamentul continuu trebuie reconsiderat cu atenție la pacienții care nu prezintă dovezi ale beneficiului terapeutic după ajustarea dozei.
Fistulizarea activă a bolii Crohn
5 mg / kg administrat sub formă de perfuzie intravenoasă, urmat de perfuzii suplimentare de 5 mg / kg în săptămâna 2 și 6 după prima perfuzie. Dacă un pacient nu răspunde după 3 doze, nu trebuie administrat tratament suplimentar cu infliximab.
La pacienții care răspund, soluțiile alternative pentru continuarea tratamentului sunt:
• Întreținere: perfuzii suplimentare de 5 mg / kg la fiecare 8 săptămâni sau
• Re-administrare: o perfuzie de 5 mg / kg dacă persistă semnele și simptomele bolii, urmată de perfuzii de 5 mg / kg la fiecare 8 săptămâni (vezi sub „Re-administrare” și pct. 4.4).
Deși lipsesc datele comparative, datele limitate la pacienții care au răspuns inițial la terapia cu 5 mg / kg, dar au pierdut răspunsul indică faptul că unii pacienți pot recupera răspunsul prin creșterea dozei (vezi pct. 5.1). Tratamentul continuu trebuie reconsiderat cu atenție la pacienții care nu prezintă dovezi ale beneficiului terapeutic după ajustarea dozei.
În boala Crohn, experiența re-administrării, dacă semnele și simptomele bolii persistă, este limitată și nu sunt disponibile date comparative de risc / beneficiu ale soluțiilor alternative pentru continuarea tratamentului.
Colită ulcerativă
O perfuzie intravenoasă de 5 mg / kg urmată de perfuzii suplimentare de 5 mg / kg la săptămânile 2 și 6 după prima perfuzie, apoi repetată la fiecare 8 săptămâni.
Datele disponibile sugerează că răspunsul clinic se obține de obicei în decurs de 14 săptămâni de la inițierea tratamentului, adică după trei administrări. Trebuie să se acorde atenție continuării tratamentului la pacienții care nu răspund în acest interval de timp.
Spondilită anchilozantă
O perfuzie intravenoasă de 5 mg / kg urmată de perfuzii suplimentare de 5 mg / kg la săptămânile 2 și 6 după prima perfuzie, apoi repetată după 6 până la 8 săptămâni. Dacă un pacient nu răspunde în decurs de 6 săptămâni (adică după 2 doze), acesta nu trebuie să primească niciun tratament suplimentar cu infliximab.
Artrita psoriazică
O perfuzie intravenoasă de 5 mg / kg urmată de perfuzii suplimentare de 5 mg / kg la săptămânile 2 și 6 după prima perfuzie, apoi repetată la fiecare 8 săptămâni.
Psoriazis
O perfuzie intravenoasă de 5 mg / kg urmată de perfuzii suplimentare de 5 mg / kg la săptămânile 2 și 6 după prima perfuzie, apoi repetată la fiecare 8 săptămâni. Dacă un pacient nu răspunde în decurs de 14 săptămâni (adică după 4 doze), nu trebuie administrat tratament suplimentar cu infliximab.
Re-administrare pentru boala Crohn și artrita reumatoidă
Dacă semnele și simptomele bolii reapar, Remicade poate fi re-administrat în termen de 16 săptămâni de la ultima perfuzie. În studiile clinice, reacțiile de hipersensibilitate întârziate au fost „mai puțin frecvente” și au apărut după intervale fără administrare de Remicade. Siguranța și eficacitatea re-administrării nu au fost stabilite după mai mult de 16 săptămâni fără administrarea Remicade. Acest lucru se aplică atât pacienților cu boală Crohn, cât și pacienților cu poliartrită reumatoidă.
Re-administrare pentru colita ulcerativă
Siguranța și eficacitatea re-administrării la alte intervale de 8 săptămâni nu au fost stabilite (vezi pct. 4.4 și 4.8).
Re-administrare pentru spondilita anchilozantă
Siguranța și eficacitatea re-administrărilor, altele decât cele administrate cu un interval de 6 până la 8 săptămâni, nu au fost stabilite (vezi pct. 4.4 și 4.8).
Re-administrare pentru artrita psoriazică
Siguranța și eficacitatea re-administrării la alte intervale de 8 săptămâni nu au fost stabilite (vezi pct. 4.4 și 4.8).
Re-administrare pentru psoriazis
O „experiență limitată în psoriazis care rezultă din re-tratament cu o singură doză de Remicade după un interval de 20 de săptămâni sugerează o„ eficacitate redusă și o „incidență mai mare a reacțiilor la perfuzie ușoare până la moderate” în comparație cu regimul inițial de inducție. pct. 5.1).
O „experiență limitată a tratamentului după agravarea bolii printr-un regim de reinducție sugerează o„ incidență ridicată a reacțiilor la perfuzie, inclusiv a celor severe, în comparație cu cele de la 8 săptămâni de tratament de întreținere (vezi punctul 4.8).
Re-administrare în diferite indicații
În cazul în care terapia de întreținere este întreruptă și este necesară reluarea tratamentului, nu se recomandă utilizarea unui regim de reinducție (vezi pct. 4.8). În această situație, tratamentul cu Remicade trebuie reluat ca doză unică urmată de doza de întreținere conform recomandărilor descrise mai sus.
Pacienți vârstnici (≥ 65 ani)
Nu s-au efectuat studii specifice cu Remicade la pacienții vârstnici. Nu au fost observate diferențe substanțiale legate de vârstă în clearance-ul sau volumul de distribuție în studiile clinice.
Nu este necesară ajustarea dozei (vezi pct. 5.2). Pentru mai multe informații despre siguranța Remicade la pacienții vârstnici, vezi pct. 4.4 și 4.8.
Insuficiență renală și / sau hepatică
Remicade nu a fost studiat la aceste populații de pacienți. Nu se poate face nicio recomandare de dozare (vezi pct. 5.2).
Populația pediatrică
Boala Crohn (6 - 17 ani)
O doză de 5 mg / kg administrată prin perfuzie intravenoasă urmată de perfuzii ulterioare de doze de 5 mg / kg la 2 și 6 săptămâni după prima perfuzie și la fiecare 8 săptămâni după aceea. Datele disponibile nu acceptă tratamentul suplimentar cu infliximab la copii și adolescenți care nu răspund în primele 10 săptămâni de tratament (vezi pct. 5.1).
Unii pacienți pot necesita un interval de doză mai scurt pentru a menține beneficiul clinic, în timp ce pentru alții poate fi suficient un interval mai mare de doză. Pacienții care au avut un interval de timp între doze redus la mai puțin de 8 săptămâni pot prezenta un risc crescut de reacții adverse. Continuarea tratamentului cu un interval scurtat trebuie luată în considerare cu atenție la acei pacienți care nu prezintă nicio dovadă de beneficiu terapeutic. intervalul de timp între doze.
Siguranța și eficacitatea Remicade la copiii cu boală Crohn sub vârsta de 6 ani nu au fost studiate.Datele farmacocinetice disponibile în prezent sunt descrise în secțiunea 5.2, dar nu se poate face nicio recomandare cu privire la posologie la copiii cu vârsta sub 6 ani.
Colită ulcerativă (6 - 17 ani)
O doză de 5 mg / kg administrată prin perfuzie intravenoasă urmată de perfuzii ulterioare de doze de 5 mg / kg la 2 și 6 săptămâni după prima perfuzie și la fiecare 8 săptămâni după aceea. Datele disponibile nu acceptă tratamentul suplimentar cu infliximab la copii și adolescenți care nu răspund în primele 8 săptămâni de tratament (vezi pct. 5.1).
Siguranța și eficacitatea Remicade la copiii cu colită ulcerativă sub vârsta de 6 ani nu au fost studiate. Datele farmacocinetice disponibile în prezent sunt descrise în secțiunea 5.2, dar nu se pot face recomandări cu privire la posologie la copii cu vârsta sub 6 ani.
Psoriazis
Siguranța și eficacitatea Remicade la copii și adolescenți cu vârsta sub 18 ani în indicația psoriazisului nu au fost stabilite. Datele disponibile în prezent sunt descrise în secțiunea 5.2, dar nu se poate face nicio recomandare cu privire la posologie.
Artrita idiopatică juvenilă, artrita psoriazică și spondilita anchilozantă
Siguranța și eficacitatea Remicade la copii și adolescenți cu vârsta sub 18 ani în indicațiile artritei idiopatice juvenile, artritei psoriazice și spondilita anchilozantă nu au fost stabilite. Datele disponibile în prezent sunt descrise în secțiunea 5.2, dar nu se poate face nicio recomandare privind o posologie.
Artrita reumatoidă juvenilă
Siguranța și eficacitatea Remicade la copii și adolescenți cu vârsta sub 18 ani în indicația artritei reumatoide juvenile nu au fost stabilite. Datele disponibile în prezent sunt descrise în secțiunile 4.8 și 5.2, dar nu se pot face recomandări privind posologia.
Insuficiență renală și / sau hepatică
Remicade nu a fost studiat la aceste populații de pacienți. Nu se poate face nicio recomandare de dozare (vezi pct. 5.2).
Mod de administrare
Remicade trebuie administrat intravenos pe o perioadă de 2 ore. Toți pacienții tratați cu Remicade trebuie observați timp de cel puțin 1-2 ore după perfuzie pentru reacții acute legate de perfuzie. Echipamentele de urgență, cum ar fi adrenalina, antihistaminicele, corticosteroizii și un aparat de respirație artificială, trebuie păstrate la dispoziție. Pacienții pot fi tratați în prealabil cu, de exemplu, un antihistaminic, hidrocortizon și / sau paracetamol, iar viteza de perfuzie poate fi redusă pentru a reduce riscul de perfuzie. reacții asociate, mai ales dacă au apărut anterior reacții legate de perfuzie (vezi pct. 4.4).
Infuzii prescurtate în indicații pentru adulți
La pacienții adulți selectați cu atenție care au tolerat cel puțin 3 perfuzii inițiale de 2 ore cu Remicade (faza de inducție) și care primesc terapie de întreținere, administrarea perfuziilor ulterioare pe o perioadă de cel puțin 1 oră Dacă o reacție de perfuzie asociată cu perfuzia scurtată se poate lua în considerare o rată de perfuzie mai lentă pentru perfuziile viitoare, în cazul în care tratamentul continuă. Perfuziile prescrise la doze> 6 mg / kg nu au fost studiate (vezi pct. 4.8).
Pentru instrucțiuni privind pregătirea și administrarea, vezi secțiunea 6.6.
04.3 Contraindicații
Pacienții cu antecedente de hipersensibilitate la infliximab (vezi pct. 4.8), la alte proteine murine sau la oricare dintre excipienții enumerați la pct. 6.1.
Pacienții cu tuberculoză sau alte infecții grave precum sepsis, abcese și infecții oportuniste (vezi pct. 4.4).
Pacienți cu insuficiență cardiacă moderată până la severă (NYHA - New York Heart Association - Clasa III / IV) (vezi pct. 4.4 și 4.8).
04.4 Avertismente speciale și precauții adecvate pentru utilizare
Pentru a îmbunătăți trasabilitatea produselor medicamentoase biologice, marca și numărul lotului produsului administrat trebuie înregistrate în mod clar (sau marcate) în evidența pacientului.
Reacții la perfuzie și hipersensibilitate
Infliximab a fost asociat cu reacții acute legate de perfuzie, inclusiv șoc anafilactic și reacții de hipersensibilitate întârziate (vezi pct. 4.8).
Reacțiile acute la perfuzie, inclusiv reacțiile anafilactice, pot apărea în timpul (în câteva secunde) sau în câteva ore după perfuzie. Dacă apar reacții acute la perfuzie, perfuzia trebuie oprită imediat. Echipamentele de urgență, cum ar fi adrenalina, antihistaminicele, corticosteroizii și un ventilator artificial, ar trebui păstrate la dispoziție Pacienții pot fi pretratați, de exemplu, cu un antihistaminic, hidrocortizon și / sau paracetamol pentru a preveni efectele ușoare și tranzitorii.
Se pot dezvolta anticorpi împotriva infliximab și au fost asociați cu o frecvență crescută a reacțiilor la perfuzie. O rată scăzută a reacțiilor la perfuzie au fost reacții alergice severe. Sa observat, de asemenea, o asociere între dezvoltarea anticorpilor la infliximab și un răspuns scăzut. Administrarea concomitentă de imunomodulatori a fost asociată cu o incidență mai mică a anticorpilor împotriva infliximabului și o reducere a frecvenței reacțiilor la perfuzie. Pacienții care au întrerupt tratamentul imunosupresor înainte sau în timpul tratamentului cu Remicade prezintă un risc crescut de a dezvolta acești anticorpi. Anticorpii împotriva infliximabului nu pot fi întotdeauna detectați în probele de ser. Dacă apar reacții severe, trebuie administrat un tratament simptomatic și nu trebuie administrate perfuzii suplimentare cu Remicade (vezi pct. 4.8).
În studiile clinice au fost raportate reacții de hipersensibilitate întârziate. Datele disponibile sugerează un risc crescut de hipersensibilitate întârziată la creșterea intervalului de timp fără administrarea Remicade. Pacienții trebuie sfătuiți să contacteze imediat medicul în cazul unui eveniment advers întârziat (vezi pct. 4.8). Dacă pacienții sunt retrași după o perioadă prelungită de timp perioadă, acestea trebuie monitorizate îndeaproape pentru semne și simptome de hipersensibilitate întârziată.
Infecții
Pacienții trebuie monitorizați îndeaproape pentru a detecta infecții, inclusiv tuberculoza, înainte, în timpul și după tratamentul cu Remicade. Deoarece eliminarea infliximab poate dura până la șase luni, monitorizarea ar trebui să continue în această perioadă. Tratamentul suplimentar cu Remicade nu trebuie administrat dacă un pacient dezvoltă infecții severe sau sepsis.
Este necesară precauție atunci când se utilizează Remicade la pacienții cu infecție cronică sau cu antecedente de infecții recurente, inclusiv terapia concomitentă cu imunosupresoare. Pacienții trebuie informați în mod corespunzător cu privire la necesitatea de a evita expunerea la potențiali factori de risc pentru infecții.
Factorul de necroză tumorală alfa (TNFα) mediază inflamația și modulează răspunsurile imune celulare. Datele experimentale demonstrează că TNFα este esențial pentru rezolvarea infecțiilor intracelulare. Experiența clinică arată că apărarea imună a gazdei este compromisă la unii pacienți tratați cu infliximab.
Trebuie remarcat faptul că supresia TNFα poate masca simptomele unei infecții, cum ar fi febra. Recunoașterea timpurie a manifestărilor clinice atipice ale infecțiilor severe și a manifestărilor clinice tipice ale infecțiilor rare și neobișnuite este esențială pentru minimizarea întârzierilor în diagnostic și tratament.
Pacienții care iau medicamente care blochează TNF sunt mai predispuși la infecții grave.
La pacienții tratați cu infliximab au fost observate tuberculoză, infecții bacteriene, inclusiv sepsis și pneumonie, fungice invazive, virale și alte infecții oportuniste. Unele dintre aceste infecții au fost fatale; cele mai frecvent raportate infecții oportuniste cu o rată a mortalității> 5% includ penumocistoză, candidoză, listerioză și aspergiloză.
Pacienții care dezvoltă o nouă infecție în timp ce sunt tratați cu Remicade trebuie să fie monitorizați cu atenție și să fie supuși unei evaluări diagnostice amănunțite. Administrarea Remicade trebuie întreruptă în cazul în care un pacient dezvoltă o nouă infecție gravă sau sepsis și se inițiază o terapie antimicrobiană sau antifungică adecvată până la rezolvarea infecției.
Tuberculoză
Au fost raportate cazuri de tuberculoză activă la pacienții tratați cu Remicade. Trebuie remarcat faptul că, în majoritatea acestor cazuri, a fost tuberculoză extrapulmonară, atât localizată, cât și difuză.
Înainte de a începe tratamentul cu Remicade, toți pacienții trebuie evaluați atât pentru tuberculoza activă, cât și pentru cea inactivă („latentă”). Această evaluare trebuie să includă un istoric medical detaliat, inclusiv un istoric personal de tuberculoză sau posibil contact anterior cu o sursă de infecție cu TBC și terapii imunosupresoare anterioare și / sau concomitente. La toți pacienții trebuie efectuate teste diagnostice adecvate, cum ar fi testul cutanat al tuberculinei și radiografia toracică (se pot aplica ghiduri locale). Se recomandă ca aceste teste să fie raportate pe cardul de alertă al pacientului. Medicii prescriptori sunt amintiți de riscul rezultatelor fals negative ale testelor cutanate la tuberculină, în special la pacienții grav bolnavi sau imunocompromiși.
Dacă este diagnosticată tuberculoza activă, nu trebuie inițiată terapia Remicade. (vezi secțiunea 4.3)
Dacă se suspectează tuberculoza latentă, trebuie consultat un medic cu experiență în tratamentul tuberculozei. În toate situațiile descrise mai jos, raportul beneficiu / risc al terapiei cu Remicade trebuie cântărit cu atenție.
Dacă este diagnosticată tuberculoza inactivă („latentă”), terapia antituberculoză pentru tuberculoza latentă trebuie începută înainte de a începe tratamentul cu Remicade în conformitate cu ghidurile locale.
La pacienții care au mulți sau factori de risc semnificativi pentru tuberculoză și au un test negativ pentru tuberculoza latentă, terapia antituberculoză trebuie luată în considerare înainte de inițierea Remicade.
Utilizarea terapiei anti-tuberculoză trebuie luată în considerare și înainte de inițierea terapiei cu Remicade la pacienții cu antecedente anterioare de tuberculoză latentă sau activă pentru care un curs adecvat de tratament nu poate fi confirmat.
Unele cazuri de tuberculoză activă au fost raportate la pacienții tratați cu Remicade în timpul și după tratamentul pentru tuberculoză latentă.
Toți pacienții trebuie sfătuiți să ceară sfatul medicului dacă apar semne / simptome sugestive de tuberculoză (de exemplu, tuse persistentă, pierderea / pierderea în greutate, febră de grad scăzut) în timpul sau după tratamentul cu Remicade.
Infecții fungice invazive
O infecție fungică invazivă, cum ar fi aspergiloză, candidoză, pneumocistoză, histoplasmoză, coccidioidomicoză sau blastomicoză, ar trebui suspectată la pacienții tratați cu Remicade dacă acestea dezvoltă boli sistemice severe și un medic competent în diagnosticarea și tratarea infecțiilor fungice invazive trebuie consultat într-un stadiu incipient. la vizitarea acestor pacienți. Infecțiile fungice invazive se pot prezenta mai degrabă ca boală diseminată decât localizată, iar testele de antigen și anticorpi pot fi negative la unii pacienți cu infecție activă. Terapia empirică antifungică adecvată trebuie luată în considerare în procesul de diagnosticare, luând în considerare atât riscul unei infecții fungice severe, cât și riscurile terapiei antifungice.
Pentru pacienții care au locuit sau au călătorit în regiuni în care infecțiile fungice invazive precum histoplasmoza, coccidioidomicoza sau blastomicoza sunt endemice, beneficiile și riscurile tratamentului cu Remicade trebuie luate în considerare cu atenție înainte de inițierea terapiei cu Remicade.
Fistulizarea bolii Crohn
Pacienții cu boală Crohn fistulizantă cu fistule supurative acute nu trebuie să inițieze terapia cu Remicade până când nu a fost exclusă o sursă de posibilă infecție, în special abcese (vezi pct. 4.3).
Reactivarea hepatitei B (VHB)
Reactivarea hepatitei B a fost observată la pacienții tratați cu un antagonist al TNF, inclusiv infliximab și care erau purtători cronici ai acestui virus. În unele cazuri, au apărut rezultate fatale.
Pacienții trebuie evaluați pentru infecția cu VHB înainte de a începe tratamentul cu Remicade.
Purtătorii cu VHB care necesită tratament cu Remicade trebuie monitorizați îndeaproape pentru semne și simptome ale infecției cu VHB activ pe toată durata terapiei și timp de câteva luni după terminarea terapiei. Nu sunt disponibile date suficiente despre pacienții cu VHB. Tratați cu terapie antivirală în asociere cu antagonistul TNF terapie pentru prevenirea reactivării VHB La pacienții care dezvoltă reactivare VHB, tratamentul cu Remicade trebuie întrerupt și trebuie inițiată o terapie antivirală eficientă cu tratament adecvat de susținere.
Evenimente hepatobiliare
În perioada de comercializare a Remicade, au fost observate cazuri foarte rare de icter și hepatită neinfecțioasă, unele cu caracteristici ale hepatitei autoimune. Au existat cazuri izolate de insuficiență hepatică care au dus la transplant hepatic sau la deces. La pacienții cu semne și simptome de disfuncție hepatică, trebuie evaluat nivelul afectării ficatului. Dacă icterul și / sau creșterea ALT ≥ 5 ori limita superioară a normalului se dezvoltă, tratamentul cu Remicade trebuie întrerupt și trebuie efectuată o examinare amănunțită a stărilor anormale.
Asocierea unui inhibitor TNF-alfa și anakinra
Infecții grave și neutropenie au apărut în studiile clinice combinate cu anakinra și un alt inhibitor al TNFα, fără niciun beneficiu clinic suplimentar asupra utilizării etanerceptului singur. Având în vedere natura evenimentelor adverse observate cu asocierea etanercept și anakinra, pot apărea toxicități similare cu combinație de anakinra și alți inhibitori de TNFα. Prin urmare, combinația de Remicade și anakinra nu este recomandată.
Asocierea unui inhibitor TNF-alfa și abatacept
În studiile clinice, utilizarea combinată de antagoniști TNF și abatacept a fost asociată cu un risc crescut de infecții, inclusiv infecții grave, comparativ cu antagoniști TNF utilizați singuri, fără o creștere a beneficiului clinic. Remicade și abatacept nu sunt recomandate.
Asocierea cu alte terapii biologice
Nu există informații suficiente cu privire la utilizarea concomitentă a infliximabului cu alte terapii biologice utilizate pentru tratarea acelorași afecțiuni ca infliximabul. Utilizarea concomitentă a infliximabului cu aceste substanțe biologice nu este recomandată din cauza posibilității unui risc crescut de infecție și a altor potențiale interacțiuni medicamentoase.
Înlocuirea între DMARD-uri biologice
Trebuie acordată precauție și pacienții trebuie monitorizați în continuare la trecerea de la un biologic la altul, deoarece activitatea biologică suprapusă poate crește și mai mult riscul de evenimente adverse, inclusiv infecție.
Vaccinuri vii / agenți terapeutici infecțioși
La pacienții tratați cu terapie anti-TNF, sunt disponibile date limitate cu privire la răspunsul la vaccinarea cu vaccinuri vii sau cu privire la transmiterea secundară a infecției cu administrarea de vaccinuri vii. Utilizarea vaccinurilor vii poate duce la infecții clinice, inclusiv infecții diseminate. . Nu se recomandă administrarea concomitentă de vaccinuri vii cu Remicade.
La sugarii expuși in utero la infliximab, după administrarea vaccinului BCG după naștere a fost raportat un rezultat fatal datorat infecției diseminate cu bacil Calmette-Guérin (BCG). Înainte de administrarea vaccinurilor vii la sugarii expuși in uter se recomandă o perioadă de așteptare de cel puțin șase luni după naștere pentru infliximab (vezi pct. 4.6).
Alte utilizări ale agenților terapeutici infecțioși, cum ar fi bacteriile vii atenuate (de exemplu, instilațiile intravesicale cu BCG pentru tratamentul cancerului) ar putea duce la infecții clinice, inclusiv infecții diseminate. Se recomandă ca agenții infecțioși terapeutici să nu fie administrați concomitent cu Remicade.
Reacții autoimune
Deficiența relativă de TNFα cauzată de terapia anti-TNF poate duce la inițierea unui proces autoimun. Dacă un pacient prezintă simptome predictive ale unui sindrom asemănător lupusului după tratamentul cu Remicade și este pozitiv pentru anticorpii anti-ADN la dubla helix, nu trebuie administrat un tratament suplimentar cu Remicade (vezi pct. 4.8).
Efecte asupra sistemului nervos
Utilizarea agenților de blocare a TNF, inclusiv infliximab, a fost asociată cu apariția sau exacerbarea simptomelor clinice și / sau dovezi radiografice ale tulburărilor demielinizante ale sistemului nervos central, incluzând scleroza multiplă și tulburări demielinizante periferice, inclusiv sindromul Guillain-Barré La pacienții cu tulburări demielinizante preexistente sau recente, beneficiile și riscurile tratamentului anti-TNF trebuie luate în considerare cu atenție înainte de a începe tratamentul cu Remicade.
Întreruperea tratamentului cu Remicade trebuie luată în considerare dacă aceste condiții se dezvoltă.
Neoplasme maligne și boli limfoproliferative
În fazele controlate ale studiilor clinice cu inhibitori de TNF, au fost observate mai multe cazuri de tumori maligne, inclusiv limfom, la pacienții care au primit un inhibitor de TNF decât la pacienții de control. În timpul studiilor clinice cu Remicade, în toate indicațiile aprobate, incidența limfomului la pacienții tratați cu Remicade a fost mai mare decât se aștepta la populația generală, dar frecvența limfomului a fost rară. În experiența de după punerea pe piață, au fost raportate cazuri de leucemie în pacienți tratați cu un antagonist al TNF. Există un risc de fond crescut de a dezvolta limfom și leucemie la pacienții cu artrită reumatoidă cu o boală inflamatorie foarte activă și de lungă durată care complică evaluarea riscului.
Într-un studiu clinic exploratoriu care a evaluat utilizarea Remicade la pacienții cu boală pulmonară obstructivă cronică moderată până la severă (Boala pulmonară obstructivă cronică, BPOC), au fost raportate mai multe cazuri de tumori maligne la pacienții tratați cu Remicade decât la pacienții de control. Toți pacienții erau fumători înrăiți. Trebuie acordată atenție evaluării tratamentului pacienților cu risc crescut de malignitate ca fumători înrăiți.
Pe baza cunoștințelor actuale, riscul apariției limfoamelor sau a tumorilor maligne la pacienții tratați cu un inhibitor TNF nu poate fi exclus (vezi pct. 4.8). Trebuie acordată atenție atunci când se ia în considerare terapia cu inhibitori de TNF la pacienții cu antecedente de malignitate sau când se ia în considerare tratamentul prelungit la pacienții care dezvoltă malignitate.
De asemenea, trebuie făcută precauție la pacienții cu psoriazis care au fost tratați anterior cu imunosupresoare sau pentru perioade prelungite cu PUVA.
În experiența ulterioară punerii pe piață, au fost raportate afecțiuni maligne, dintre care unele letale, la copii, adolescenți și adulți tineri (până la 22 de ani) tratați cu medicamente care blochează TNF (inițierea tratamentului ≤ 18 ani), inclusiv Remicade. jumătate din cazuri au fost limfoame, celelalte cazuri au fost o varietate de malignități diferite și au inclus malignități rare, de obicei asociate cu imunosupresia. Nu poate fi exclus un risc pentru apariția neoplasmelor maligne la pacienții tratați cu inhibitori de TNF.
Au fost raportate cazuri rare după punerea pe piață a limfomului cu celule T hepatosplenice (HSTCL) la pacienții tratați cu agenți de blocare a TNF, inclusiv infliximab. Această formă rară de limfom cu celule T are un curs și un rezultat extrem de agresiv. pacienții au primit tratament cu AZA sau 6-MP concomitent cu sau imediat înainte de un blocant TNF. Marea majoritate a cazurilor cu Remicade au apărut la pacienții cu boala Crohn sau colită ulcerativă și Majoritatea cazurilor au fost raportate la adolescenți sau bărbați tineri. trebuie luat în considerare cu atenție riscul potențial al combinației de AZA sau 6-MP și Remicade. Nu poate fi exclus un risc de apariție a limfomului cu celule T hepatosplenice la pacienții tratați cu Remicade (vezi pct. 4.8).
Melanomul și carcinomul celular Merkel au fost raportate la pacienții care au primit tratament cu un blocant TNF, inclusiv Remicade (vezi pct. 4.8). Se recomandă examinarea periodică a pielii, în special la pacienții cu factori de risc de cancer de piele.
Un studiu retrospectiv de cohortă bazat pe date din registrele naționale de sănătate suedeze a constatat o incidență crescută a cancerului de col uterin la femeile cu poliartrită reumatoidă tratate cu infliximab comparativ cu pacienții netratați biologic sau cu populația generală, inclusiv cu cei cu vârsta peste 60 de ani. tratat cu Remicade, inclusiv cei cu vârsta peste 60 de ani.
Toți pacienții cu colită ulcerativă care prezintă un risc crescut de a dezvolta displazie de colon sau carcinom (de exemplu, pacienții cu colită ulcerativă de lungă durată sau colangită sclerozantă primară) sau care au antecedente medicale de displazie sau cancer de colon ar trebui investigați. la această displazie la intervale regulate, înainte de începerea terapiei și în cursul bolii. Această evaluare ar trebui să includă o colonoscopie și biopsii în conformitate cu orientările locale.În lumina datelor actuale nu se știe dacă tratamentul cu infliximab afectează riscul de a dezvolta displazie sau cancer de colon (vezi pct. 4.8).
Deoarece nu a fost stabilită posibilitatea unui risc crescut de a dezvolta cancer la pacienții tratați cu Remicade cu displazie nou diagnosticată, este necesar să se evalueze raportul beneficiu / risc la pacienții individuali și să se ia în considerare întreruperea tratamentului.
Insuficienta cardiaca
Remicade trebuie utilizat cu precauție la pacienții cu insuficiență cardiacă ușoară (clasa I / II NYHA). Pacienții trebuie monitorizați îndeaproape și tratamentul cu Remicade trebuie întrerupt la pacienții care prezintă simptome noi sau agravante ale insuficienței cardiace (vezi pct. 4.3 și 4.8).
Reacții hematologice
Au fost raportate cazuri de pancitopenie, leucopenie, neutropenie și trombocitopenie la pacienții tratați cu medicamente anti-TNF, inclusiv Remicade. Toți pacienții trebuie sfătuiți să solicite asistență medicală imediată dacă dezvoltă semne sau simptome compatibile ale discraziei sanguine (de exemplu, febră persistentă, vânătăi, sângerări și paloare). Întreruperea tratamentului cu Remicade trebuie luată în considerare la pacienții cu anomalii hematologice semnificative confirmate.
Alții
Experiența cu siguranța tratamentului cu Remicade la pacienții care au suferit o intervenție chirurgicală, inclusiv artroplastia, este limitată. Timpul de înjumătățire prin eliminare lung al infliximab trebuie luat în considerare la planificarea intervenției chirurgicale. Un pacient care necesită intervenție chirurgicală în timpul tratamentului cu Remicade trebuie monitorizat îndeaproape pentru un risc crescut de infecții și trebuie luate în considerare măsurile adecvate.
Nerespectarea tratamentului pentru boala Crohn poate indica prezența stricturilor fibroase rigide care pot necesita tratament chirurgical. Nu există dovezi clinice care să sugereze că infliximab se agravează sau provoacă stricturi fibrotice.
Populații speciale
Pacienți vârstnici (≥ 65 ani)
Incidența infecțiilor grave la pacienții cu vârsta de 65 de ani și peste tratați cu Remicade a fost mai mare decât la pacienții cu vârsta sub 65 de ani. Unii dintre aceștia au fost letali. O atenție deosebită trebuie acordată riscului de infecție la tratarea vârstnicilor (vezi pct. 4.8) .
Populația pediatrică
Infecții
În studiile clinice, infecțiile au fost raportate mai frecvent la copii și adolescenți decât la populațiile adulte (vezi pct. 4.8).
Vaccinări
Se recomandă ca pacienții copii și adolescenți, dacă este posibil, să facă toate vaccinările în conformitate cu cele mai recente linii directoare înainte de inițierea tratamentului cu Remicade.
Neoplasme maligne și tulburări limfoproliferative
În experiența ulterioară punerii pe piață, au fost raportate afecțiuni maligne, dintre care unele letale, la copii, adolescenți și adulți tineri (până la 22 de ani) tratați cu medicamente care blochează TNF (inițierea tratamentului ≤ 18 ani), inclusiv Remicade. jumătate din cazuri au fost limfoame, celelalte cazuri au fost o varietate de malignități diferite și au inclus malignități rare, de obicei asociate cu imunosupresia. Nu poate fi exclus un risc pentru apariția tumorilor maligne la copii și adolescenți tratați cu inhibitori de TNF.
Cazuri rare de limfom cu celule T hepatosplenice au fost raportate după punerea pe piață la pacienții tratați cu agenți de blocare a TNF, inclusiv infliximab. Această formă rară de limfom cu celule T are un curs extrem de agresiv și de obicei rezultat fatal. Aproape toți pacienții au primit tratament cu AZA sau 6-MP concomitent cu sau imediat înainte de un blocant TNF. Marea majoritate a cazurilor cu Remicade au apărut la pacienții cu boala Crohn sau colită ulcerativă și majoritatea cazurilor au fost raportate la adolescenți sau bărbați tineri. combinația dintre AZA sau 6-MP și Remicade trebuie luată în considerare cu atenție. Nu poate fi exclus un risc de apariție a limfomului cu celule T hepatosplenice la pacienții tratați cu Remicade (vezi pct. 4.8).
04.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu au fost efectuate studii de interacțiune.
Există indicații că utilizarea concomitentă a metotrexatului și a altor imunomodulatori la pacienții cu poliartrită reumatoidă, artrită psoriazică și boala Crohn reduce formarea de anticorpi împotriva infliximab și crește concentrațiile plasmatice de infliximab. Cu toate acestea, rezultatele sunt incerte datorită limitărilor metodelor utilizate pentru testarea infliximabului și a anticorpilor împotriva infliximabului în ser.
Corticosteroizii nu par să modifice farmacocinetica infliximabului într-un mod relevant clinic.
Nu se recomandă asocierea Remicade cu alte terapii biologice utilizate pentru tratarea acelorași afecțiuni ca Remicade, inclusiv anakinra și abatacept (vezi pct. 4.4).
Se recomandă ca vaccinurile vii să nu fie administrate în același timp cu Remicade. De asemenea, se recomandă ca vaccinurile vii să nu fie administrate sugarilor după expunere in uter la infliximab timp de cel puțin 6 luni după naștere (vezi pct. 4.4).
Agenții terapeutici infecțioși nu trebuie administrați concomitent cu Remicade (vezi pct. 4.4).
04.6 Sarcina și alăptarea
Femeile aflate la vârsta fertilă
Femeile aflate la vârsta fertilă trebuie să utilizeze contracepție adecvată în timpul tratamentului cu Remicade și să continue utilizarea acestuia timp de cel puțin 6 luni după ultima doză.
Sarcina
Un număr moderat de date colectate prospectiv asupra pacienților gravide (aproximativ 450) tratați cu infliximab cu rezultate cunoscute, inclusiv un număr limitat (aproximativ 230) de sarcini tratate în primul trimestru, nu au prezentat efecte neașteptate asupra rezultatului. Datorită inhibării TNFα, infliximab administrat în timpul sarcinii poate modifica răspunsurile imune normale ale nou-născutului. Nici o toxicitate maternă, embriotoxicitate și teratogenitate nu au fost găsite într-un studiu de toxicitate asupra dezvoltării la șoareci, folosind un anticorp similar care inhibă selectiv funcționalitatea TNFα (vezi secțiunea 5.3).
Experiența clinică disponibilă este prea limitată pentru a exclude riscurile și, prin urmare, administrarea infliximab nu este recomandată în timpul sarcinii.
Infliximab trece prin placentă și a fost detectat în serul sugarilor până la 6 luni după naștere. După expunere in uter la infliximab, sugarii pot avea un risc mai mare de infecție, inclusiv o „infecție severă diseminată care poate avea un rezultat fatal. Administrarea de vaccinuri vii (de exemplu, vaccin BCG) la sugarii expuși in uter infliximab nu este recomandat timp de cel puțin 6 luni după naștere (vezi pct. 4.4 și 4.5). De asemenea, au fost raportate cazuri de agranulocitoză (vezi pct. 4.8).
Timp de hrănire
Nu se știe dacă infliximab este excretat în laptele matern sau absorbit sistemic după ingestie. Deoarece imunoglobulinele umane sunt excretate în laptele matern, femeile nu trebuie să alăpteze cel puțin 6 luni după tratamentul cu Remicade.
Fertilitate
Sunt disponibile date preclinice insuficiente pentru a trage concluzii cu privire la efectele infliximabului asupra fertilității și funcției generale de reproducere (vezi pct. 5.3).
04.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Remicade are efecte minore asupra capacității de a conduce vehicule sau de a folosi utilaje. Pot apărea amețeli după administrarea Remicade (vezi pct. 4.8).
04.8 Efecte nedorite
Rezumatul profilului de siguranță
Infecția tractului respirator superior a fost cea mai frecventă reacție adversă (ADR) raportată în studiile clinice, care a apărut la 25,3% dintre pacienții tratați cu infliximab comparativ cu 16,5% dintre pacienții martor. "Utilizarea inhibitorilor TNF raportați pentru Remicade include reactivarea VHB, insuficiență cardiacă congestivă ( CHF), infecții severe (inclusiv sepsis, infecții oportuniste și TBC), boală serică (reacții de hipersensibilitate întârziată), reacții hematologice, lupus sistemic eritematos / sindrom asemănător lupusului, boală demielinizantă, evenimente hepatobiliare, limfom, HSTCL, leucemie, carcinom cu celule Merkel , melanom, malignitate pediatrică, sarcoidoză / reacție de tip sarcoid, abces intestinal sau perianal (în boala Crohn) și reacții severe la perfuzie (vezi pct. 4.4).
Tabelul reacțiilor adverse
Tabelul 1 enumeră reacțiile adverse raportate în studiile clinice, precum și reacțiile adverse, unele cu rezultate fatale, raportate după punerea pe piață. În cadrul sistemului de organe, reacțiile adverse sunt enumerate în funcție de frecvență, utilizând următoarele categorii: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 până la
tabelul 1
Efecte nedorite în studiile clinice și după punerea pe piață
* inclusiv tuberculoza bovină (infecție BCG diseminată), vezi pct. 4.4
Reacții legate de perfuzie
În studiile clinice, o reacție legată de perfuzie a fost definită ca orice eveniment advers care apare în timpul perfuziei sau în decurs de 1 oră după perfuzie. În studiile clinice de fază III, 18% dintre pacienții tratați cu infliximab comparativ cu 5% dintre pacienții tratați cu placebo au avut o reacție legată de perfuzie. În general, o proporție mai mare de pacienți care au primit monoterapie cu infliximab au prezentat o reacție legată de perfuzie decât pacienții care au primit concomitent infliximab cu imunomodulatori. Aproximativ 3% dintre pacienți au întrerupt tratamentul din cauza reacțiilor legate de perfuzie și toți pacienții s-au recuperat cu sau fără terapie medicală.
Dintre pacienții tratați cu infliximab care au avut o reacție la perfuzie în timpul perioadei de inducție până la săptămâna 6, 27% au prezentat o reacție la perfuzie în perioada de întreținere cuprinsă între săptămâna 7 și săptămâna 54. Din pacienții care nu au avut o reacție la perfuzie în timpul perioadei de inducție, 9% au prezentat o reacție la perfuzie în perioada de întreținere.
Într-un studiu clinic efectuat la pacienți cu poliartrită reumatoidă (ASPIRE), perfuziile au fost administrate timp de 2 ore pentru primele 3 perfuzii. infuzia. În acest studiu, șaizeci și șase la sută dintre pacienți (686 din 1040) au primit cel puțin o perfuzie scurtată cu o durată de 90 de minute sau mai puțin și 44% dintre pacienți (454 din 1040) au primit cel puțin o perfuzie scurtată cu o durată de 60 de minute sau mai puțin. La pacienții tratați cu infliximab care au primit cel puțin o perfuzie scurtată, reacții legate de perfuzie au apărut la 15% dintre pacienți, iar reacții severe la perfuzie au apărut la 0,4% dintre pacienți.
Într-un studiu clinic efectuat la pacienți cu boala Crohn (SONIC), reacții legate de perfuzie au fost observate la 16,6% (27/163) dintre pacienții care au primit monoterapie cu infliximab, la 5% (9/179) dintre pacienții care au primit monoterapie cu infliximab. în asociere cu AZA și la 5,6% (9/161) dintre pacienții cărora li sa administrat monoterapie AZA. O reacție severă la perfuzie (
În perioada de după punerea pe piață, cazurile de reacții anafilactoide, inclusiv edem laringian / faringian, bronhospasm sever și convulsii, au fost asociate cu administrarea Remicade.
În plus, au existat, de asemenea, raportări rare de pierdere tranzitorie a vederii și ischemie miocardică / infarct miocardic care au apărut în timpul sau în decurs de două ore de la perfuzia cu Remicade (vezi pct. 4.4).
Reacții la perfuzie după re-administrarea Remicade
Un studiu clinic a fost conceput la pacienții cu psoriazis moderat până la sever pentru a evalua eficacitatea și siguranța terapiei de întreținere pe termen lung, comparativ cu tratamentul cu un regim de inducție Remicade (maximum patru perfuzii la 0, 2, 6 și 14 săptămâni) după boală Pacienții nu au primit tratament imunosupresor concomitent. În brațul de tratament, 4% (8/219) dintre pacienți au prezentat reacții severe la perfuzie față de edem facial și hipotensiune arterială. În toate cazurile, tratamentul cu Remicade a fost oprit și / sau un alt tratament a fost adoptat cu rezolvarea completă a semnelor și simptomelor.
Hipersensibilitate întârziată
În studiile clinice, reacțiile de hipersensibilitate întârziate au fost mai puțin frecvente și au apărut după intervale de timp fără Remicade mai mici de 1 an. În studiile de psoriazis, reacțiile de hipersensibilitate întârziate au apărut devreme în timpul tratamentului. Semnele și simptomele includ mialgie și / sau artralgie cu febră și / sau erupții cutanate, cu unii pacienți care prezintă mâncărime, edem facial, de mână sau de buze, disfagie, urticarie, dureri în gât și cefalee.
Sunt disponibile date insuficiente cu privire la incidența reacțiilor de hipersensibilitate întârziate după intervale de timp fără Remicade mai mari de 1 an, dar datele limitate din studiile clinice sugerează un risc crescut de hipersensibilitate întârziată la creștere. Durata intervalelor de timp fără administrarea Remicade (vezi pct. 4.4).
Într-un studiu clinic de 1 an cu perfuzii repetate la pacienții cu boala Crohn (studiu ACCENT I), incidența reacțiilor rezultate din dezvoltarea reacțiilor asemănătoare bolii serice a fost de 2,4%.
Imunogenitate
Pacienții care au dezvoltat anticorpi împotriva infliximabului au fost mai predispuși să aibă reacții legate de perfuzie (de aproximativ 2 până la 3 ori mai mari).
În studiile clinice în care s-au administrat doze unice și multiple de infliximab variind de la 1 la 20 mg / kg, anticorpi împotriva infliximab au fost găsiți la 14% dintre pacienții care au primit orice tratament imunosupresor și la 24% dintre pacienții fără tratament imunosupresor. 8% dintre pacienții cu poliartrită reumatoidă tratați în mod repetat cu doza recomandată și cu metotrexat au dezvoltat anticorpi împotriva infliximabului. și 26% dintre pacienții care nu au primit metotrexat la momentul inițial). La pacienții cu boala Crohn care au primit tratament de întreținere în medie 3,3% dintre pacienții care au primit imunosupresoare și 13,3% dintre pacienții care nu au primit imunosupresori au dezvoltat anticorpi împotriva infliximabului. Incidența anticorpilor a fost de 2-3 de ori mai mare pentru pacienții tratați episodic. Datorită limitărilor metodologice, un test negativ nu a exclus prezența anticorpilor împotriva infliximabului. Unii pacienți care au dezvoltat titruri ridicate de anticorpi împotriva infliximab au avut eficacitate redusă. La pacienții cu psoriazis tratați cu regim de întreținere infliximab, în absența tratamentului concomitent cu imunomodulatori, aproximativ 28% au dezvoltat anticorpi împotriva infliximab (vezi pct. 4.4: „Reacții la perfuzie și hipersensibilitate”).
Infecții
Tuberculoza, infecțiile bacteriene, inclusiv sepsisul și pneumonia, fungice invazive, virale și alte infecții oportuniste au fost observate la pacienții cărora li s-a administrat Remicade. Unele dintre acestea au fost fatale. Cele mai frecvent raportate infecții oportuniste cu o rată de mortalitate> 5% includ pneumocistoză, candidoză, listerioză și aspergiloză (vezi pct. 4.4).
În studiile clinice, 36% dintre pacienții tratați cu infliximab au fost tratați pentru infecții, comparativ cu 25% dintre pacienții tratați cu placebo.
În studiile clinice privind poliartrita reumatoidă, incidența infecțiilor grave, inclusiv pneumonia, a fost mai mare la pacienții tratați cu infliximab și metotrexat decât la cei tratați numai cu metotrexat, în special la doze de 6 mg / kg sau mai mari (vezi pct. 4.4).
Dintre rapoartele spontane raportate în perioada de după punerea pe piață, infecțiile sunt cel mai frecvent eveniment advers grav. Unele dintre cazuri au avut un rezultat fatal. Aproape 50% din decesele raportate au fost asociate cu infecția. Au fost raportate cazuri de tuberculoză., uneori letale, inclusiv cazuri de tuberculoză miliară și tuberculoză de localizare extrapulmonară (vezi pct. 4.4).
Neoplasme maligne și boli limfoproliferative
În studiile clinice efectuate cu infliximab în care au fost tratați 5.780 de pacienți, reprezentând 5.494 de ani-pacient, au fost detectate 5 cazuri de limfoame și 26 de cazuri de malignitate non-limfom, comparativ cu niciun caz de limfom și 1 caz de malignitate non-limfom. observat la 1.600 de pacienți tratați cu placebo, reprezentând 941 de ani-pacient.
În studiile de siguranță pe termen lung de până la 5 ani cu infliximab, reprezentând 6.234 pacienți-ani (3.210 pacienți), au fost raportate 5 cazuri de limfom și 38 de cazuri de tumori maligne non-limfom.
De asemenea, au fost raportate cazuri de tumori maligne, inclusiv limfom, în perioada de după punerea pe piață (vezi pct. 4.4).
Într-un studiu clinic explorator care a implicat pacienți cu BPOC moderată până la severă care au fost fie fumători, fie foști fumători, 157 pacienți adulți au fost tratați cu Remicade la doze similare cu cele utilizate în artrita reumatoidă și boala Crohn. Nouă dintre acești pacienți au dezvoltat tumori maligne, inclusiv 1 limfom. Durata mediană a unei monitorizări a fost de 0,8 ani (incidență 5,7% [IÎ 95% 2,65% - 10,6%]. Un caz de malignitate a fost raportat la cei 77 de pacienți din grupul de control (durata mediană de urmărire 0,8 ani; incidență 1,3% [IÎ 95% 0,03% - 7,0%]). Majoritatea acestor afecțiuni maligne au implicat plămâni, cap sau gât.
Un studiu de cohortă retrospectivă a populației a constatat o incidență crescută a cancerului de col uterin la femeile tratate cu infliximab cu poliartrită reumatoidă comparativ cu pacienții netratați biologic sau cu populația generală, inclusiv cu cei cu vârsta peste 60 de ani (vezi pct. 4.4).
În plus, cazuri rare de limfom cu celule T hepatosplenice au fost raportate după punerea pe piață la pacienții tratați cu Remicade, marea majoritate a cazurilor au apărut la pacienții cu boala Crohn și colită ulcerativă, majoritatea pacienților fiind adolescenți sau bărbați tineri (vezi pct. 4.4). ).
Insuficienta cardiaca
Într-un studiu de fază II cu scopul de a evalua Remicade în insuficiența cardiacă congestivă (CHF), o incidență mai mare a mortalității datorată agravării insuficienței cardiace a fost găsită la pacienții tratați cu Remicade, în special la cei tratați cu cea mai mare doză de 10 mg / kg. (adică dublul dozei maxime aprobate). În acest studiu, 150 de pacienți cu CHF NYHA clasa III și IV (fracție de ejecție a ventriculului stâng ≤ 35%), au fost tratați cu 3 perfuzii de Remicade 5 mg / kg, 10 mg / kg sau placebo pe o perioadă de 6 săptămâni. La 38 de săptămâni, 9 din cei 101 pacienți tratați cu Remicade (2 până la 5 mg / kg și 7 până la 10 mg / kg) au murit în timp ce a existat un deces în rândul celor 49 de pacienți tratați cu placebo.
Au fost raportate cazuri de agravare a insuficienței cardiace, cu sau fără factori de declanșare identificabili, în perioada post-introducere pe piață la pacienții tratați cu Remicade. nicio boală cardiovasculară preexistentă cunoscută Unii dintre acești pacienți aveau vârsta sub 50 de ani.
Evenimente hepatobiliare
În studiile clinice, au fost observate creșteri ușoare sau moderate ale ALT și AST la pacienții cărora li s-a administrat Remicade fără a evolua până la leziuni hepatice severe. Au fost observate creșteri ale ALT ≥ 5 x peste limitele normale (ULN) (vezi Tabelul 2). Creșteri ale aminotransferazei (mai frecvente în ALT decât AST) au fost observate la o proporție mai mare de pacienți cărora li s-a administrat Remicade decât în grupurile de control, atât atunci când Remicade a fost administrat singur, cât și când a fost administrat în asociere cu alte medicamente imunosupresoare. Majoritatea anomaliilor aminotransferazei au fost tranzitorii; cu toate acestea, au apărut creșteri prelungite la un număr mic de pacienți. În general, pacienții care au dezvoltat creșteri ale ALT și AST au fost asimptomatice, iar anomaliile au scăzut sau s-au rezolvat fie prin continuarea sau întreruperea tratamentului cu Remicade, fie prin schimbarea terapiei concomitente. Au fost raportate cazuri foarte rare de icter și hepatită, unele cu caracteristici ale hepatitei autoimune, la pacienții cărora li s-a administrat Remicade în perioada de supraveghere după punerea pe piață (vezi pct. 4.4).
masa 2
Numărul de pacienți cu activitate ALT crescută în studiile clinice
1 Pacienții din grupul placebo au primit metotrexat, în timp ce pacienții din grupul cu infliximab au primit atât infliximab, cât și metotrexat.
2 pacienți din grupul placebo din cele 2 studii de boală Crohn de fază III, ACCENT I și ACCENT II, au primit o doză inițială de 5 mg / kg infliximab la inițierea studiului și placebo în faza de întreținere. Au fost randomizați la grupul de întreținere placebo și ulterior au trecut la infliximab, au fost incluși în grupul infliximab în analiza ALT. În studiul de fază IIIb asupra bolii Crohn, SONIC, pacienții din brațul placebo au primit AZA 2,5 mg / kg / zi ca control activ, pe lângă perfuziile cu infliximab placebo.
3 Numărul de pacienți evaluați pentru ALT.
4 Urmărirea medie se bazează pe pacienții tratați.
Anticorpi antinucleari (ANA) / anticorpi cu ADN dublu catenar (ADNd)
Aproximativ jumătate dintre pacienții tratați cu infliximab din studiile clinice care au fost ANA negative la momentul inițial au devenit ANA pozitivi în timpul studiului, comparativ cu aproximativ o cincime din pacienții tratați cu placebo. Anticorpii anti-dsDNA au fost detectați recent la aproximativ 17% dintre pacienții tratați cu infliximab, comparativ cu 0% dintre pacienții tratați cu placebo. În cea mai recentă evaluare, 57% dintre pacienții tratați cu infliximab au rămas pozitivi pentru anticorpii anti-ADNs.
Populația pediatrică
Pacienți cu artrită reumatoidă juvenilă
Remicade a fost studiat într-un studiu clinic care a implicat 120 de pacienți (interval de vârstă: 4-17 ani) cu poliartrită reumatoidă activă juvenilă, indiferent de utilizarea metotrexatului. Pacienții au fost tratați cu infliximab 3 sau 6 mg / kg ca regim de inducție cu 3 doze (săptămâna 0 , 2, 6 sau săptămâna 14,16, respectiv 20) urmată de terapie de întreținere la fiecare 8 săptămâni, în asociere cu metotrexat.
Reacții la perfuzie
Reacțiile la perfuzie au apărut la 35% dintre pacienții cu artrită reumatoidă juvenilă cărora li s-au administrat 3 mg / kg, comparativ cu 17,5% dintre pacienții cărora li s-a administrat 6 mg / kg. o posibilă reacție anafilactică (dintre care 2 au fost incluse în reacțiile severe la perfuzie). În grupul cu 6 mg / kg, 2 din 57 de pacienți au prezentat o reacție severă la perfuzie. ).
Imunogenitate
38% dintre pacienții cărora li s-a administrat 3 mg / kg au dezvoltat anticorpi împotriva infliximab, comparativ cu 12% dintre pacienții care au primit 6 mg / kg. Titrurile de anticorpi au fost semnificativ mai mari în grupul care a primit 3 mg / kg decât în grupul care a primit 6 mg / kg.
Infecții
Infecțiile au apărut la 68% (41/60) dintre copiii care au primit 3 mg / kg timp de 52 de săptămâni, la 65% (37/57) dintre copiii care au primit 6 mg / kg infliximab timp de 38 de săptămâni și la 47% (28 / 60) dintre copiii cărora li s-a administrat placebo timp de 14 săptămâni (vezi pct. 4.4).
Pacienți copii cu boala Crohn
Următoarele reacții adverse au fost raportate mai frecvent la pacienții copii cu boală Crohn incluși în studiul REACH (vezi pct. 5.1) decât la pacienții adulți cu boala Crohn: anemie (10,7%), sânge în scaun (9,7%), leucopenie (8,7%), înroșirea feței cu roșeață a pielii (8,7%), infecții virale (7,8%), neutropenie (6,8%), fracturi osoase (6,8%), infecții bacteriene (5,8%) și reacții alergice care implică tractul respirator (5,8%). Alte considerații speciale sunt prezentate mai jos.
Reacții legate de perfuzie
17,5% dintre pacienții randomizați din studiul REACH au prezentat 1 sau mai multe reacții la perfuzie. Nu au fost raportate cazuri grave de reacții la perfuzie și 2 subiecți din studiul REACH au dezvoltat reacții anafilactice neserioase.
Imunogenitate
Anticorpi împotriva infliximab au fost detectați la 3 (2,9%) dintre pacienții copii.
Infecții
În studiul REACH, infecțiile au fost raportate la 56,3% dintre subiecții randomizați tratați cu infliximab. Infecțiile au fost raportate mai frecvent la subiecții cărora li s-au administrat perfuzii la fiecare 8 săptămâni decât la cei tratați la fiecare 12 săptămâni (73,6% și respectiv 38,0%), în timp ce infecții grave au fost raportate la 3 subiecți din grupul de tratament de întreținere la fiecare 8 săptămâni și la 4 subiecți în grupul tratat la fiecare 12 săptămâni. Cele mai frecvent raportate infecții au fost infecțiile tractului respirator superior și faringita. Abcesul a fost cel mai frecvent dintre infecțiile grave.3 au fost raportate 3 cazuri de pneumonie (1 severă) și 2 cazuri de herpes zoster (ambele non-grave).
Pacienți copii cu colită ulcerativă
În general, reacțiile adverse raportate în studiu la copii și adolescenți cu colită ulcerativă (C0168T72) au fost în general consistente cu cele raportate în studii la pacienții adulți cu colită ulcerativă (ACT 1 și ACT 2). În studiul C0168T72, cele mai frecvente reacții adverse au fost infecția tractului respirator superior, faringita, durerea abdominală, febra și cefaleea. Cel mai frecvent eveniment advers a fost o agravare a colitei ulcerative, a cărei incidență a fost mai mare la pacienții tratați la fiecare 12 săptămâni decât la cei tratați la fiecare 8 săptămâni.
Reacții legate de perfuzie
În general, 8 (13,3%) din 60 de pacienți tratați au raportat una sau mai multe reacții la perfuzie, cu 4 din 22 de pacienți (18,2%) în grupul de întreținere la fiecare 8 săptămâni și 3 din 23 de pacienți (13, 0%) din grupul de întreținere tratați la fiecare 12 săptămâni Nu au fost raportate reacții grave la perfuzie. Toate reacțiile la perfuzie au fost ușoare sau moderate ca intensitate.
Imunogenitate
Anticorpii împotriva infliximabului au fost detectați la 4 (7,7%) dintre pacienți până în săptămâna 54.
Infecții
Infecțiile au fost raportate la 31 (51,7%) din cei 60 de pacienți tratați în studiul C0168T72 și 22 (36,7%) au necesitat tratament antimicrobian oral sau parenteral. Proporția pacienților cu infecții în studiul C0168T72 a fost similară cu cea din studiul pediatric privind boala Crohn (REACH), dar mai mare decât proporția în studiile privind colita ulcerativă la adulți (ACT 1 și ACT 2). Incidența generală a infecțiilor în studiul C0168T72 a fost de 13/22 (59%) în grupul de întreținere tratat la fiecare 8 săptămâni și de 14/23 (60,9%) în grupul de întreținere tratat la fiecare 12 săptămâni. Căile respiratorii superioare (7/60 [12] %]) și faringita (5/60 [8%]) au fost cele mai frecvent raportate infecții ale sistemului respirator. Infecții grave au fost raportate la 12% (7/60) din toți pacienții tratați.
În acest studiu, au existat mai mulți pacienți în grupul de vârstă de la 12 la 17 ani decât în grupul de vârstă de la 6 la 11 ani (45/60 [75,0%]) față de 15/60 [25,0%]).Deși numărul pacienților din fiecare subgrup a fost prea mic pentru a face concluzii ferme cu privire la „efectul vârstei asupra evenimentelor legate de siguranță”, a existat o proporție mai mare de pacienți cu evenimente adverse grave și întreruperea tratamentului cauzată de evenimente. grupul de vârstă mai tânăr decât în grupul de vârstă mai mare. Deși proporția pacienților cu infecții a fost, de asemenea, mai mare în grupul de vârstă mai mică, pentru infecțiile severe, proporțiile din cele două grupuri au fost similare. Proporția generală a evenimentelor adverse și a reacțiilor la perfuzie similar între grupele de vârstă 6-11 ani și 12-17 ani.
Experiență post-marketing
Raportarea spontană după punerea pe piață a evenimentelor adverse grave la copii și adolescenți a inclus tumori maligne, inclusiv limfom cu celule T hepatosplenice, anomalii tranzitorii ale enzimelor hepatice, sindroame asemănătoare lupusului și autoanticorpi pozitivi (vezi pct. 4.4 și 4.8).
Informații suplimentare despre populații speciale
Pacienți vârstnici (≥ 65 ani)
În studiile clinice privind artrita reumatoidă, incidența infecțiilor grave a fost mai mare la pacienții tratați cu infliximab plus metotrexat cu vârsta de 65 de ani și peste (11,3%) decât la pacienții cu vârsta sub 65 de ani (4, 6%). La pacienții tratați numai cu metotrexat, incidența infecțiilor grave a fost de 5,2% la pacienții cu vârsta de 65 de ani și peste, comparativ cu 2,7% la pacienții cu vârsta sub 65 de ani (vezi pct. 4.4).
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate care apar după autorizarea medicamentului este importantă, deoarece permite monitorizarea continuă a raportului beneficiu / risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată la Agenția italiană pentru medicamente. , site-ul web: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Supradozaj
Nu au fost raportate cazuri de supradozaj. S-au administrat doze unice până la maximum 20 mg / kg fără efecte toxice.
05.0 PROPRIETĂȚI FARMACOLOGICE
05.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: inhibitori ai factorului de necroză tumorală alfa (TNF-alfa).
Codul ATC: L04AB02.
Mecanism de acțiune
Infliximab este un anticorp monoclonal himeric, murin-uman, care se leagă cu afinitate ridicată atât la formele solubile, cât și transmembranare ale TNFα, dar nu la limfotoxina α (TNFβ).
Efecte farmacodinamice
Infliximab inhibă in vitro Activitatea TNFα într-o gamă largă de doze biologice. Infliximab a prevenit boala la șoarecii transgenici care dezvoltă poliartrită ca o consecință a expresiei esențiale a TNFα umană și, atunci când a fost administrat după debutul bolii, a permis regresia eroziunilor articulare. In vivo, infliximab formează rapid complexe stabile cu TNFα uman, proces care duce la pierderea activității biologice a TNFα.
Concentrații mari de TNFα au fost detectate la articulațiile pacienților cu poliartrită reumatoidă și legate de activitatea ridicată a bolii. Tratamentul cu infliximab a dus la o reducere a infiltrării celulelor inflamatorii în zonele inflamate ale articulațiilor și la reducerea expresiei în poliartrita reumatoidă, molecule care mediază aderența celulelor, chimiotaxia și degradarea țesuturilor. După tratamentul cu infliximab, pacienții au prezentat niveluri serice reduse de interleukină 6 (IL-6) și proteine C reactive (CRP) și niveluri ridicate de hemoglobină la pacienții cu RA cu niveluri reduse de hemoglobină comparativ cu valorile de pre-tratament. În plus, limfocitele din sângele periferic nu au prezentat o scădere semnificativă a numărului și a răspunsului proliferativ la test in vitro de stimulare mitogenică comparativ cu celulele pacienților netratați. La pacienții cu psoriazis, tratamentul cu infliximab a dus la scăderea inflamației epidermice și la normalizarea diferențierii keratinocitelor în plăci psoriazice.În artrita psoriazică, tratamentul pe termen scurt cu Remicade a redus numărul de celule T și vase de sânge.
O evaluare histologică a biopsiilor de colon efectuată înainte și la 4 săptămâni după administrarea infliximab a relevat o reducere substanțială a TNFα detectabil. Tratamentul cu Infliximab la pacienții cu boală Crohn a fost, de asemenea, asociat cu un substanțial
scăderea concentrației serice a CRP, un marker inflamator frecvent crescut. Numărul total de leucocite periferice a fost minim afectat la pacienții tratați cu infliximab, deși modificările limfocitelor, monocitelor și neutrofilelor au reflectat modificări față de valorile normale. Celulele mononucleare din sângele periferic (PBMC) ale pacienților tratați cu infliximab au prezentat o capacitate de răspuns proliferativ neafectată la stimuli, comparativ cu pacienții netratați; în plus, după tratamentul cu infliximab, nu au fost observate modificări substanțiale în producția de citokine de către celulele PBMC stimulate. Analiza celulelor mononucleare ale laminei proprii obținută în urma biopsiei mucoasei intestinale a arătat că tratamentul cu infliximab a dus la o reducere a numărului de celule capabile să exprime TNFα și interferon γ. Alte studii histologice au furnizat dovezi că tratamentul cu infliximab reduce infiltrarea celulelor inflamatorii în zonele implicate ale intestinului și prezența marcatorStudiile endoscopice ale mucoasei intestinale au arătat vindecarea mucoasei la pacienții tratați cu infliximab.
Eficacitate și siguranță clinică
Artrită reumatoid la adulți
Eficacitatea infliximab a fost evaluată în două studii clinice pilot multicentric, randomizate, dublu-orb: ATTRACT și ASPIRE. În ambele studii, utilizarea concomitentă a dozelor stabile de acid folic, corticosteroizi orali (≤ 10 mg / die) și / sau antiinflamatoare steroidiene (AINS).
Obiectivele principale au fost reducerea semnelor și simptomelor definite de criteriile Colegiului American de Reumatologie (ACR20 pentru ATTRACT, indicator ACR-N pentru ASPIRE), prevenirea deteriorării structurale a articulațiilor și îmbunătățirea funcției fizice. Simptomele au fost definite ca o îmbunătățire de cel puțin 20% (ACR20) în numărul articulațiilor dureroase și umflate și în 3 din următoarele 5 criterii: evaluarea globală a medicului, evaluarea globală a pacientului, evaluarea funcției / dizabilității, scala vizuală analogică a durerii, rata de sedimentare a eritrocitelor sau C- proteină reactivă. ACR-N utilizează aceleași criterii ca ACR20, calculat luând în considerare cel mai mic procent de îmbunătățire a numărului de articulații umflate, articulații dureroase și mediana celor 5 componente rămase ale răspunsului ACR. Deteriorarea articulației structurale (eroziune și reducerea liniei articulare) la ambele mâini și picioare a fost măsurată prin evaluarea schimbării față de linia de bază în scorul total Sharp modificat de van der Heijde (0-440). Chestionarul de evaluare a sănătății (HAQ; scara 0 la 3) a fost utilizat pentru a evalua schimbarea medie în timp față de valoarea inițială a funcției fizice.
Studiul ATTRACT a evaluat răspunsurile la săptămânile 30, 54 și 102 într-un studiu controlat cu placebo la 428 pacienți cu poliartrită reumatoidă activă în ciuda tratamentului cu metotrexat. Aproximativ 50% dintre pacienți se aflau în clasa funcțională III. Pacienții au fost tratați cu placebo, 3 mg / kg sau 10 mg / kg infliximab în săptămânile 0, 2 și 6 și la fiecare 4 sau 8 săptămâni după aceea. Toți pacienții au luat o doză stabilă de metotrexat (mediană de 15 mg / săptămână) timp de 6 luni înainte de înscriere și au rămas în doze stabile pe tot parcursul studiului.
Rezultatele din săptămâna 54 (ACR20, scor total Sharp modificat de van der Heijde și HAQ), sunt prezentate în Tabelul 3. O incidență mai mare a răspunsului clinic (ACR50 și ACR70) a fost observată în toate grupurile tratate cu infliximab în săptămânile 30 și 54 comparativ cu metotrexatul singur.
O reducere a ratei de progresie a deteriorării structurale a articulațiilor (eroziune și reducerea decalajului articular) a fost observată la toate grupurile tratate cu infliximab la 54 de săptămâni (Tabelul 3).
Efectele observate în săptămâna 54 au fost menținute până în săptămâna 102 de tratament. Datorită numărului de întreruperi ale tratamentului, amploarea diferenței de efect între grupurile de monoterapie cu infliximab și metotrexat nu a putut fi definită.
Tabelul 3
Efecte asupra ACR20, deteriorarea articulațiilor structurale și funcția fizică în săptămâna 54, ATTRACT
controlat = Toți pacienții au avut RA activă în ciuda tratamentului cu doze stabile de metotrexat timp de 6 luni înainte de înscriere și au trebuit să rămână la doze stabile în timpul studiului. Utilizarea concomitentă a dozelor stabile de corticosteroizi orali (≤ 10 mg / zi) și / sau medicamentul antiinflamator nesteroidian (AINS) a fost permis; s-a dat un supliment de folat.
b Toate dozele de infliximab au fost administrate concomitent cu metotrexat și folat și, în unele cazuri, cu corticosteroizi și / sau antiinflamatoare nesteroidiene (AINS)
c p
d valori mai mari indică leziuni articulare mai mari.
și HAQ = Chestionar de evaluare a sănătății; valori mai mari indică un „handicap mai mic.
Studiul ASPIRE a evaluat răspunsurile la săptămâna 54 la 1004 de pacienți naivi anterior cu metotrexat cu poliartrită reumatoidă activă (număr mediu al articulațiilor umflate și sensibile: 19 și, respectiv, 31) de debut recent (durata bolii ≤ 3 ani, mediană de 0,6 ani). Toți pacienții au primit metotrexat (optimizat la 20 mg / săptămână până la săptămâna 8) în asociere cu placebo sau infliximab 3 mg / kg sau 6 mg / kg în săptămânile 0, 2 și 6 și la fiecare 8 săptămâni după aceea. Rezultatele din săptămâna 54 sunt prezentate în Tabelul 4.
După 54 de săptămâni de tratament, ambele doze de infliximab + metotrexat au dat o îmbunătățire semnificativă statistic semnificativă a semnelor și simptomelor decât metotrexatul singur, măsurată prin proporția pacienților care au obținut răspunsuri ACR 20, 50 și 70.
În ASPIRE, mai mult de 90% dintre pacienți au avut cel puțin două radiografii evaluabile. Reducerea ratei de progresie a leziunilor structurale a fost observată la săptămânile 30 și 54 în grupurile infliximab + metotrexat comparativ cu metotrexatul singur.
Tabelul 4
Efecte asupra ACRn, deteriorarea articulațiilor structurale și funcția fizică în săptămâna 54, ASPIRE
pe p
b valori mai mari indică o deteriorare articulară mai mare.
c chestionar de evaluare a stării de sănătate; valori mai mari indică un „handicap mai mic.
d p = 0,030 e
Datele care susțin titrarea dozei în artrita reumatoidă provin din studii
ATRACT, ASPIRE și START. START a fost un studiu randomizat, multicentric, dublu-orb, cu 3 brațe, paralel, studiu de siguranță. Într-una din brațele studiului (grupul 2, n = 329), pacienților cu răspuns inadecvat li s-a permis titrarea dozei în trepte de 1,5 mg / kg de la 3 la 9 mg / kg. Majoritatea (67%) dintre acești pacienți nu au necesitat titrarea dozei. Dintre pacienții care l-au solicitat, 80% au obținut răspuns clinic și majoritatea (64%) dintre aceștia au necesitat doar o creștere de 1,5 mg / kg.
Boala Crohn la adulți
Tratamentul de inducție în boala Crohn activă, moderată până la severă Eficacitatea unei doze unice de infliximab a fost evaluată la 108 pacienți cu boală Crohn activă, variind în mărime de la moderată la severă (indicele de activitate al bolii Crohn (CDAI) ≥ 220 ≤ 400) într-un studiu randomizat, dublu-orb, controlat cu placebo. Dintre cei 108 pacienți, 27 au fost tratați cu doza recomandată de infliximab (5 mg / kg). Toți pacienții au avut un răspuns inadecvat la terapiile convenționale anterioare A fost permisă utilizarea concomitentă a dozelor nemodificate de terapii convenționale și 92% dintre pacienți au continuat primiți astfel de terapii.
Obiectivul principal a fost calculul numărului de pacienți care au prezentat un răspuns clinic, definit ca o scădere a CDAI ≥ 70 puncte față de momentul inițial, în săptămâna 4 și fără creșterea utilizării medicamentelor sau a intervențiilor chirurgicale pentru boală. Pacienții care au răspuns în săptămâna 4 au fost urmăriți până în săptămâna 12. Obiectivele secundare au inclus numărul de pacienți în remisie clinică în săptămâna 4 (CDAI
În săptămâna 4, după administrarea unei doze unice, 22/27 (81%) pacienți tratați cu infliximab la o doză de 5 mg / kg au avut un răspuns clinic comparativ cu 4/25 (16%) pacienți tratați cu placebo (p
Tratament de întreținere în boala Crohn activă, moderată până la severă la adulți
Eficacitatea perfuziilor repetate cu infliximab a fost evaluată într-un studiu clinic de un an (ACCENT I). Un total de 573 pacienți cu boală Crohn activă moderată până la severă (CDAI ≥ 220 ≤ 400) au primit o singură perfuzie de 5 mg / kg pe săptămână 0. 178 din cei 580 de pacienți înrolați (30,7%) au avut boală severă (scor CDAI> 300 și tratament concomitent cu corticosteroizi și / sau imunosupresoare) corespunzător populației definite în indicații (vezi pct. 4.1). În săptămâna 2, toți pacienții au fost evaluați pentru răspuns clinic și randomizat într-unul din cele 3 grupuri de tratament; un grup de întreținere placebo, un grup de întreținere de 5 mg / kg și o întreținere cu 10 mg / kg Apoi, toate cele 3 grupuri au primit perfuzii repetate în săptămâna 2, 6 și la fiecare 8 săptămâni după aceea .
Dintre cei 573 de pacienți randomizați, 335 (58%) au obținut răspuns clinic în săptămâna 2. Acești pacienți au fost clasificați ca respondenți în săptămâna 2 și incluși în analiza primară (vezi Tabelul 5). grupul de întreținere placebo și 42% (68/163) din grupul de întreținere cu infliximab au obținut răspuns clinic la săptămâna 6. După aceea, nu a existat nicio diferență între grupuri în numărul de pacienți care au răspuns ulterior la terapie.
Punctele finale co-primare au fost procentul de pacienți în remisie clinică (CDAI
Tabelul 5
Efecte asupra vitezei de răspuns și remisie, date de la ACCENT I (pacienții care au răspuns la săptămâna 2)
a Reducerea CDAI ≥ 25% și ≥ 70 puncte.
b CDAI
La începutul săptămânii 14, pacienții care au răspuns la tratament, dar și-au pierdut beneficiul clinic, au fost trecuți la o doză de infliximab cu 5 mg / kg mai mare decât doza la care au fost inițial randomizați. L "Optzeci și nouă la sută (50 / 56) dintre pacienții care au pierdut răspunsul clinic la terapia de întreținere cu 5 mg / kg infliximab după săptămâna 14 au răspuns la 10 mg / kg tratament cu infliximab.
La săptămânile 30 și 54, s-au observat îmbunătățiri în evaluările calității vieții, o reducere a spitalizărilor legate de boală și utilizarea corticosteroizilor în grupurile de întreținere cu infliximab comparativ cu grupul de întreținere placebo.
Infliximab, cu sau fără AZA, a fost evaluat într-un studiu randomizat, dublu-orb, comparator activ (SONIC) la 508 pacienți adulți cu boală Crohn moderată până la severă (CDAI ≥ 220 ≤ 450) care nu au mai fost tratați până acum. imunosupresoare și cu o durată mediană a bolii de 2,3 ani. La momentul inițial, 27,4% dintre pacienți erau tratați cu corticosteroizi sistemici, 14,2% dintre pacienții tratați cu budesonidă și 54,3% dintre pacienții cu compuși 5-ASA. Pacienții au fost randomizați pentru a primi monoterapie AZA, monoterapie cu infliximab sau infliximab plus terapie combinată AZA. Infliximab a fost administrat la o doză de 5 mg / kg la săptămânile 0, 2, 6 și la fiecare 8 săptămâni după aceea. AZA a fost administrat la o doză zilnică de 2,5 mg / kg.
Obiectivul principal al studiului a fost remisia clinică fără corticosteroizi în săptămâna 26, definită ca pacienți în remisiune clinică (prednison CDAI sau echivalent) sau budesonidă în doză> 6 mg / zi. Pentru rezultate, vezi Tabelul 6. Proporțiile pacienților cu vindecare mucoasă în săptămâna 26 au fost semnificativ mai mari în grupurile combinate cu infliximab plus AZA (43,9%, p
Tabelul 6
Procentul pacienților care au obținut remisie clinică fără corticosteroizi în săptămâna 26, SONIC
* Valorile P reprezintă fiecare grup de tratament cu infliximab comparativ cu monoterapia cu AZA
Modele similare în realizarea remisiei clinice fără corticosteroizi au fost observate în săptămâna 50. În plus, s-a observat o îmbunătățire a calității vieții cu infliximab, așa cum a fost raportat de chestionarul IBDQ.
Tratamentul de inducție în boala Crohn fistulizantă activă
Eficacitatea a fost evaluată într-un studiu randomizat, dublu-orb, controlat cu placebo la 94 de pacienți cu boala Crohn care au avut fistule timp de cel puțin 3 luni. Treizeci și unu dintre acești pacienți au fost tratați cu 5 mg / kg de infliximab. Aproximativ 93% dintre pacienți au fost supuși anterior terapiei cu antibiotice sau imunosupresoare.
A fost permisă utilizarea concomitentă, nemodificată, a terapiilor convenționale și 83% dintre pacienți au continuat să primească cel puțin una dintre aceste terapii. Pacienții au primit trei doze de placebo sau infliximab în săptămânile 0, 2 și 6. Urmărirea pacienților a fost de 26 săptămâni Obiectivul primar a fost numărul de pacienți care au prezentat un răspuns clinic, definit ca o reducere ≥ 50% față de valoarea inițială a numărului de fistule de purjare după compresie ușoară în cel puțin două controale consecutive (4 săptămâni mai târziu), fără creșterea consumului de droguri. sau chirurgie pentru boala Crohn.
68% (21/31) dintre pacienții cărora li sa administrat infliximab în doză de 5 mg / kg au prezentat un răspuns clinic comparativ cu 26% (8/31) dintre pacienții tratați cu placebo (p = 0,002). Timpul mediu de răspuns la grupul cu infliximab a fost de 2 săptămâni. Durata medie a răspunsului a fost de 12 săptămâni. În plus, închiderea tuturor fistulelor a fost observată la 55% dintre pacienții care au primit infliximab, comparativ cu 13% dintre pacienții cărora li s-a administrat placebo (p = 0,001).
Tratamentul de întreținere în boala Crohn fistulizantă activă
Eficacitatea perfuziilor repetate de infliximab la pacienții cu boală Crohn fistulizantă a fost evaluată într-un studiu de 1 an (ACCENT II). Un total de 306 pacienți au primit 3 doze de 5 mg / kg infliximab în săptămânile 0, 2 și 6. La momentul inițial , 87% dintre pacienți au avut fistule perianale, 14% au avut fistule abdominale, 9% au avut fistule rectovaginale. Scorul CDAI median a fost de 180. În săptămâna 14, 282 de pacienți au fost evaluați pentru răspunsul clinic și au fost randomizați pentru a fi tratați cu placebo sau infliximab 5 mg / kg la fiecare 8 săptămâni până la săptămâna 46.
Pacienții care au răspuns în săptămâna 14 (195/282) au fost analizați pentru obiectivul primar, care a fost timpul dintre randomizare și pierderea răspunsului (vezi tabelul 7). Scăderea corticosteroizilor a fost permisă după săptămâna 6.
Tabelul 7
Efecte asupra vitezei de răspuns, date din studiul ACCENT II (pacienții care au răspuns la săptămâna 14)
o reducere de ≥ 50% față de valoarea inițială a numărului de fistule drenante pe o perioadă de ≥ 4 săptămâni
b Absența fistulelor drenante
La începutul săptămânii 22, pacienții care au răspuns inițial la tratament și care au pierdut răspunsul au fost trecuți la tratamentul activ la fiecare 8 săptămâni la o doză de infliximab cu 5 mg / kg mai mare decât doza la care fuseseră randomizați inițial. grupul de întreținere cu 5 mg / kg infliximab care a trecut la tratamentul activ deoarece au pierdut răspunsul la reducerea fistulei după săptămâna 22, 57% (12/21) au răspuns la tratamentul infliximab 10 mg / kg la fiecare 8 săptămâni.
Nu a existat nicio diferență semnificativă între placebo și infliximab în proporția pacienților cu închidere susținută a tuturor fistulelor până în săptămâna 54, în simptomele proctalgiei, abcesului și infecțiilor tractului urinar sau în numărul de fistule noi dezvoltate în timpul tratamentului.
Terapia de întreținere cu infliximab la fiecare 8 săptămâni a redus semnificativ spitalizările și intervențiile chirurgicale legate de boală în comparație cu placebo. În plus, s-a observat o reducere a utilizării corticosteroizilor și o îmbunătățire a calității vieții.
Colita ulcerativă la adulți
Siguranța și eficacitatea Remicade au fost evaluate în două studii clinice randomizate, dublu-orb, controlate cu placebo (ACT 1 și ACT 2) la pacienți adulți cu colită ulcerativă activă moderată până la severă (scor Mayo 6-12; sub-scor endoscopic ≥ 2) cu răspuns inadecvat la terapiile convenționale [corticosteroizi orali, aminosalicilați și / sau imunomodulatori (6 MP, AZA)]. A fost permisă administrarea concomitentă de doze fixe de aminosalicilați orali, corticosteroizi și / sau medicamente imunolodulatoare. Pacienții din studii au fost randomizați pentru a primi placebo sau Remicade 5 mg / kg sau Remicade 10 mg / kg la săptămânile 0, 2, 6, 14 și 22 și în ACT 1 la săptămânile 30, 38 și 46. Reducerea corticosteroizilor a fost permisă după 8 săptămâni.
Tabelul 8
Efecte asupra răspunsului clinic, remisiei clinice și vindecării mucoasei la săptămânile 8 și 30.
Date combinate din ACT 1 și 2
pe p
Eficacitatea Remicade în săptămâna 54 a fost evaluată în studiul ACT 1.
În săptămâna 54, 44,9% dintre pacienții din grupul combinat cu infliximab au avut un răspuns clinic comparativ cu 19,8% din grupul placebo (p
O proporție mai mare de pacienți din grupul combinat cu infliximab au reușit să întrerupă tratamentul cu corticosteroizi și să rămână în remisie clinică comparativ cu grupul placebo la ambele săptămâni 30 (22,3% față de 7,2%, p
Datele combinate din studiile ACT 1 și ACT 2 și extensiile acestora, analizate de la momentul inițial până în săptămâna 54, au demonstrat o reducere a spitalizărilor legate de colită ulcerativă și a intervențiilor chirurgicale după tratamentul cu infliximab. Numărul spitalizărilor legate de colita ulcerativă a fost semnificativ mai mic în grupurile de tratament cu infliximab 5 și 10 mg / kg comparativ cu grupul placebo (numărul mediu de spitalizări la 100 subiecți pe an: 21 și 19 versus 40 în grupul placebo; p = 0,019 și, respectiv, p = 0,007).
Numărul de intervenții chirurgicale legate de colita ulcerativă a fost, de asemenea, mai mic în grupurile de tratament cu infliximab 5 și 10 mg / kg comparativ cu grupul placebo (numărul mediu de intervenții chirurgicale la 100 subiecți pe an: 22 și 19 versus 34; p = 0.145 și p = 0,022, respectiv).
Numărul subiecților care au suferit colectomie în orice moment în cursul celor 54 de săptămâni după prima perfuzie a agentului de studiu a fost colectat și combinat cu date din studiile ACT 1 și ACT 2 și extensiile acestora. Mai puțini subiecți au fost supuși colectomiei în infliximab 5 mg / kg (28/242 sau 11,6% [NS]) și în grupul cu infliximab 10 mg / kg (18/242 sau 7,4% [p = 0,011]) comparativ cu grupul placebo (36/244; 14,8%).
Reducerea incidenței colectomiilor a fost, de asemenea, examinată într-un alt studiu randomizat, dublu-orb (C0168Y06) la pacienți spitalizați (n = 45) cu colită ulcerativă activă moderată până la severă, care nu au reușit să răspundă la corticosteroizii intravenoși. risc crescut de colectomie. Au existat semnificativ mai puține colectomii în decurs de 3 luni de la perfuzie la pacienții cărora li s-a administrat o doză unică de 5 mg / kg infliximab comparativ cu pacienții cărora li s-a administrat placebo (29,2%, respectiv 66,7%, p = 0,017).
În studiile ACT 1 și ACT 2, infliximab a îmbunătățit calitatea vieții, confirmată de o îmbunătățire semnificativă statistic atât a măsurii unui parametru specific al bolii, IBDQ, cât și în îmbunătățirea celor 36 de întrebări generice care alcătuiesc SF-36.
Spondilita anchilozantă la adulți
Eficacitatea și siguranța infliximabului au fost studiate în două studii multicentric dublu-orb, controlate cu placebo, la pacienți cu spondilită anchilozantă activă (scorul Indexului de baie al activității bolii spondilitei anchilozante [BASDAI] scor ≥ 4 și durere spinală ≥ 4 pe o scară de la 1 la 10).
În primul studiu (P01522), care a inclus o fază dublu-orbă de 3 luni, 70 de pacienți au fost tratați fie cu infliximab 5 mg / kg, fie cu placebo în săptămânile 0, 2, 6 (35 pacienți pe grup). Din săptămâna 12, pacienții tratați până acum cu placebo au început să primească infliximab în doză de 5 mg / kg la fiecare 6 săptămâni până la săptămâna 54. După primul an, 53 de pacienți au fost plasați într-un protocol deschis până în săptămâna 102.
Într-un al doilea studiu clinic (ASSERT), 279 de pacienți au fost randomizați la tratament cu placebo (grupul 1, n = 78) sau infliximab 5 mg / kg (grupul 2, n = 201) la săptămânile 0, 2, 6 și la fiecare 6 săptămâni până la săptămâna 24. După aceea, toți subiecții studiați au continuat tratamentul cu infliximab la fiecare 6 săptămâni până la săptămâna 96. Grupul 1 a primit o doză de 5 mg / kg de infliximab. În grupul 2, începând cu săptămâna 36, pacienții care au avut un BASDAI ≥ 3 pentru 2 vizite consecutive au fost tratați cu o doză de infliximab de 7,5 mg / kg la fiecare 6 săptămâni până la săptămâna 96.
În studiul ASSERT, îmbunătățirea semnelor și simptomelor a fost observată din săptămâna 2. În săptămâna 24, numărul pacienților care au avut un răspuns ASAS 20 a fost de 15/78 (19%) în grupul placebo și egal cu 123/201 (61 %) în grupul infliximab 5 mg / kg (p
În studiul P01522, îmbunătățirea semnelor și simptomelor a fost observată din săptămâna 2. În săptămâna 12, pacienții care au avut un răspuns BASDAI 50 au fost 3/35 (9%) în grupul placebo și 20/35 (57%) în infliximab 5 grupa mg / kg (p
În ambele studii, funcția fizică și calitatea vieții, măsurate de BASFI și scorul componentelor fizice pe scara SF-36, s-au îmbunătățit semnificativ.
Artrita psoriazică la adulți
Eficacitatea și siguranța au fost evaluate în două studii dublu-orb, controlate cu placebo, multicentrice, la pacienți cu artrită psoriazică activă.
În primul studiu clinic (IMPACT), eficacitatea și siguranța infliximabului au fost studiate la 104 pacienți cu artrită psoriazică activă poliarticulară. În timpul fazei dublu-orb de 16 săptămâni, pacienții au primit fie 5 mg / kg de infliximab, fie palcebo. 0, 2, 6 și 14 (52 de pacienți din fiecare grup). Începând cu săptămâna 16, pacienții din grupul placebo au fost trecuți la infliximab și apoi toți pacienții au primit infliximab la o doză de 5 mg / kg fiecare. 8 săptămâni până la săptămână 46. După primul an al studiului, 78 de pacienți au continuat cu o extensie deschisă până în săptămâna 98.
În al doilea studiu clinic (IMPACT 2), eficacitatea și siguranța infliximabului au fost studiate la 200 de pacienți cu artrită psoriazică activă (articulații umflate ≥ 5 și articulații dureroase ≥ 5). Patruzeci și șase la sută dintre pacienți au continuat cu doze fixe de metotrexat ( ≤ 25 mg / săptămână) În timpul fazei dublu-orb de 24 de săptămâni, pacienții au primit fie 5 mg / kg de infliximab, fie placebo în săptămânile 0, 2, 6, 14 și 22 (100 de pacienți din fiecare grup). 47 de pacienți au primit placebo cu îmbunătățire
Rezultatele cheie ale eficacității pentru IMPACT și IMPACT 2 sunt prezentate mai jos
Tabelul 9
Efecte asupra ACR și PASI în IMPACT și IMPACT 2
* Analiza ITT în care au fost incluși subiecții cu date lipsă Nu-respondenți
în săptămâna 98 datele IMPACT includ pacienții din grupul placebo și pacienții tratați cu infliximab care au intrat în extensia deschisă
b Pe baza pacienților cu PASI ≥ 2,5 la momentul inițial pentru IMPACT și a pacienților cu implicare a suprafeței corporale psoriazice (ASB) la momentul inițial ≥ 3% în IMPACT 2
** Răspunsul PASI 75 pentru IMPACT nu este inclus din cauza nivelului scăzut de N; p
În IMPACT și IMPACT 2, răspunsul clinic a fost observat încă din săptămâna 2 și a fost menținut până în săptămâna 98 și, respectiv, în săptămâna 54. Eficacitatea a fost demonstrată cu și fără utilizarea concomitentă de metotrexat. La pacienții tratați cu infliximab au fost observate scăderi ale parametrilor activității periferice caracteristice artritei psoriazice (cum ar fi numărul articulațiilor umflate, numărul articulațiilor dureroase / sensibile, dactilită și prezența entezopatiilor).
Modificările radiografice au fost evaluate în IMPACT2. Radiografiile mâinilor și picioarelor au fost colectate la momentul inițial, săptămâna 24 și săptămâna 54. Tratamentul cu Infliximab a redus rata de progresie a afectării articulațiilor periferice comparativ cu tratamentul placebo la punctul final primar din săptămâna 24, măsurată ca schimbare față de valoarea inițială a scorului total modificat vdH -S (scorul mediu ± SD a fost de 0,82 ± 2,62 în grupul placebo comparativ cu - 0,70 ± 2,53 în grupul cu infliximab; p
La pacienții tratați cu infliximab s-a demonstrat o îmbunătățire semnificativă a funcției fizice, evaluată prin HAQ, precum și îmbunătățiri semnificative ale calității vieții măsurate prin scorul sumar al componentelor fizice și mentale ale SF-36 în IMPACT 2.
Psoriazis la adulți
Eficacitatea infliximab a fost evaluată în două studii randomizate, dublu-orb, multicentrice, SPIRIT și EXPRESS. Pacienții din ambele studii au avut psoriazis în plăci (BSA [Suprafața corpului] ≥ 10% și scor PASI [Zona psoriazisului și Indicele de severitate] ≥ 12 ) Obiectivul primar în ambele studii a fost proporția pacienților care au obținut o îmbunătățire ≥ 75% față de valoarea inițială a scorului PASI în săptămâna 10.
SPIRIT a evaluat eficacitatea terapiei de inducție a infliximab la 249 de pacienți cu psoriazis în plăci tratați anterior cu PUVA sau terapie sistemică. Pacienții au primit perfuzii de 3 sau 5 mg / kg de infliximab sau placebo în săptămânile 0, 2 și 6. Pacienții cu PGA ≥ 3 au fost eligibili pentru a primi o perfuzie suplimentară cu același tratament în săptămâna 26.
În SPIRIT, proporția pacienților care au obținut PASI 75 în săptămâna 10 a fost de 71,7% în grupul cu infliximab 3 mg / kg, de 87,9% în grupul cu infliximab 5 mg / kg și în grupul de infliximab 5 mg / kg. 5,9% în grupul grup placebo (p 20 săptămâni. Nu s-au observat fenomene de revenire.
EXPRESS a evaluat eficacitatea terapiei de inducție și de întreținere cu infliximab la 378 de pacienți cu psoriazis în placă. Pacienții au primit 5 mg / kg infliximab sau perfuzii placebo în săptămânile 0, 2 și 6 urmate de fiecare 8 terapii de întreținere. Săptămâni până în săptămâna 22 în grupul placebo și până în săptămâna 46 în grupul cu infliximab. În săptămâna 24, grupul placebo a trecut la terapia de inducție cu infliximab (5 mg / kg), urmată de terapia de întreținere cu infliximab (5 mg / kg). ) Terapia anterioară cu PUVA, metotrexat, ciclosporină sau acitretină a fost primită de 71,4% dintre pacienți, deși acestea nu au fost neapărat rezistente la terapie. Cele mai semnificative rezultate sunt prezentate în Tabelul 10. La subiecții tratați cu infliximab, răspunsurile semnificative la PASI 50 au fost evident la primul v isita (săptămâna 2) și răspunsuri la PASI 75 la a doua vizită (săptămâna 6). Eficacitatea în subgrupul de pacienți care au urmat anterior terapii sistemice a fost similară cu cea a populației generale studiate.
Tabelul 10
Rezumatul răspunsurilor PASI, răspunsurile PGA și procentul de pacienți cu toate unghiile vindecate în săptămânile 10, 24 și 50. EXPRESS.
pe p
b n = 292
c Analiza a fost efectuată la subiecții cu psoriazis al unghiilor la momentul inițial (81,8% dintre subiecți).
Îmbunătățiri semnificative față de valoarea inițială au fost evidente în DLQI (p
Populația pediatrică
Boala Crohn la copii și adolescenți (6-17 ani)
În studiul REACH, 112 pacienți (cu vârsta cuprinsă între 6-17 ani, vârsta medie 13 ani) cu boală Crohn activă moderată până la severă (media CDAI pediatrică de 40) și cu un răspuns inadecvat la terapiile convenționale au fost tratați cu 5 mg / kg infliximab la săptămânile 0, 2 și 6. O doză stabilă de 6-MP, AZA sau MTX a fost necesară pentru toți pacienții (35% au fost, de asemenea, pe corticosteroizi la momentul inițial). Pacienții considerați de investigator că au răspuns clinic în săptămâna 10 au fost apoi randomizați în două grupuri și au primit infliximab 5 mg / kg la fiecare 8 săptămâni sau la fiecare 12 săptămâni ca terapie de întreținere. Dacă s-a pierdut răspunsul în timpul întreținerii, s-a permis trecerea la o doză mai mare (10 mg / kg) și / sau la intervale mai scurte între perfuzii (la fiecare 8 săptămâni). Treizeci și doi de pacienți pediatrici evaluabili în scopul studiului au suferit această tranziție (9 subiecți din grupul tratați la fiecare 8 săptămâni și 23 de subiecți din grupul tratați la fiecare 12 săptămâni). Douăzeci și patru dintre acești pacienți (75,0%) și-au recăpătat un răspuns clinic după această schimbare.
Procentul pacienților cu răspuns clinic în săptămâna 10 a fost de 88,4% (99/112). Procentul de subiecți care au obținut remisie clinică în săptămâna 10 a fost de 58,9% (66/112).
În săptămâna 30, procentul pacienților în remisie clinică a fost mai mare în grupul la fiecare 8 săptămâni (59,6%, 31/52) decât cel al pacienților din grupul de întreținere la fiecare 12 săptămâni (35,3%, 18/51; p = 0,013) . În săptămâna 54, datele au fost după cum urmează: 55,8% (29/52) în grupul de întreținere tratat la fiecare 8 săptămâni și 23,5% (12/51) în grupul de întreținere tratat la fiecare 12 săptămâni (p
Datele despre fistule au fost extrase din scorurile PCDAI. Dintre cei 22 de pacienți care au avut fistule la momentul inițial, 63,6% (14/22), 59,1% (13/22) și 68,2% (15/22) au răspuns complet la fistula. La săptămânile 10, 30 și 54, respectiv, luând în considerare, în general, grupurile de întreținere atât la cei tratați la fiecare 8 săptămâni, cât și la cei tratați la fiecare 12 săptămâni.
În plus, a fost observată o îmbunătățire semnificativă statistic și clinic a calității vieții și a înălțimii, precum și o reducere semnificativă a utilizării corticosteroizilor de la momentul inițial.
Colita ulcerativă pediatrică (6-17 ani)
Siguranța și eficacitatea infliximab au fost evaluate într-un studiu clinic multicentric, randomizat, deschis, paralel (C0168T72) la 60 de copii și adolescenți cu vârsta cuprinsă între 6-17 ani (vârsta mediană 14,5 ani) cu colită. Mayo scor 6-12; subscore endoscopice ≥ 2) cu răspuns inadecvat la terapiile convenționale. La momentul inițial 53% dintre pacienți primeau terapie imunomodulatoare (6-MP, AZA și / sau MTX) și 62% dintre pacienți primeau corticosteroizi. imunomodulatorii și reducerea conicității corticosteroizilor au fost permise după săptămâna 0.
Toți pacienții au primit un regim de inducție a infliximab 5 mg / kg în săptămânile 0, 2 și 6. Pacienții care nu au răspuns la infliximab în săptămâna 8 (n = 15) nu au primit medicamente și s-au întors pentru o evaluare a siguranței. În săptămâna 8, 45 de pacienți au fost randomizați și au primit tratament de întreținere cu infliximab 5 mg / kg la fiecare 8 săptămâni sau la fiecare 12 săptămâni.
Proporția pacienților cu răspuns clinic în săptămâna 8 a fost de 73,3% (44/60). Răspunsul clinic în săptămâna 8 a fost similar între pacienții cu și fără utilizarea concomitentă de imunomodulatori la momentul inițial. Remisiunea clinică în săptămâna 8 a fost de 33,3% (17/51), măsurată prin scorul indicelui de activitate al colitei ulcerative pediatrice (PUCAI).
În săptămâna 54, proporția pacienților în remisie clinică măsurată prin scorul PUCAI a fost de 38% (8/21) în grupul de întreținere de 8 săptămâni și de 18% (4/22) în fiecare grup de întreținere de 12 săptămâni. . Pentru pacienții care au primit corticosteroizi la momentul inițial, proporția pacienților în remisie și care nu au primit corticosteroizi în săptămâna 54 a fost de 38,5% (5/13) în grupul de întreținere la fiecare 8 săptămâni și 0% (0/13) în grupul de întreținere tratat la fiecare 12 săptămâni.
În acest studiu, au existat mai mulți pacienți în grupul de vârstă 12 - 17 ani decât în grupul de vârstă 6-11 ani (45/60 vs. 15/60). Deși numărul pacienților din fiecare subgrup era prea mic pentru a trage concluzii ferme cu privire la „efectul vârstei”, a existat un număr mai mare de pacienți din grupul de vârstă mai tânăr care au crescut doza sau au întrerupt tratamentul datorită eficacității inadecvate.
Alte indicații pediatrice
Agenția Europeană a Medicamentului a renunțat la obligația de a prezenta rezultatele studiilor cu Remicade la toate subseturile populației pediatrice în artrita reumatoidă, artrita idiopatică juvenilă, artrita psoriazică, spondilita anchilozantă, psoriazisul și boala Crohn (a se vedea secțiunea 4.2 pentru informații privind utilizarea la copii și adolescenți) ).
05.2 "Proprietăți farmacocinetice
Perfuziile intravenoase unice de 1, 3, 5, 10 sau 20 mg / kg de infliximab au crescut atât concentrația plasmatică maximă (Cmax), cât și zona de sub curba concentrație-timp (ASC) într-o manieră proporțională cu doza. (Vd median 3,0-4,1 litri) a fost independent de doza administrată, arătând astfel că infliximab este distribuit în principal în compartimentul vascular. Nu s-a observat dependența de timp a caracteristicilor farmacocinetice. infliximab nu a fost caracterizat. Infliximab nemodificat nu a fost recuperat în urină. Nu diferențe majore în clearance-ul sau volumul de distribuție legate de vârstă sau greutate au fost observate la pacienții cu poliartrită reumatoidă. Farmacocinetica infliximab la pacienții vârstnici nu a fost studiată. Nu au fost efectuate studii la pacienții cu insuficiență hepatică sau renală.
La doze unice de 3, 5 sau 10 mg / kg, valorile medii ale Cmax au fost de 77, 118 și respectiv 277 mcg / ml. Timpul mediu de înjumătățire plasmatică la aceste doze a variat între 8 și 9,5 zile. La majoritatea pacienților, la doza unică recomandată de 5 mg / kg pentru boala Crohn și 3 mg / kg la fiecare 8 săptămâni pentru întreținere. În artrita reumatoidă, infliximab ar putea să fie detectat în ser timp de cel puțin 8 săptămâni.
Administrarea repetată de infliximab (5 mg / kg la săptămânile 0, 2 și 6 în boala Crohn fistulizantă, 3 sau 10 mg / kg la 4 sau 8 săptămâni în artrita reumatoidă) a dus la o ușoară acumulare de infliximab în ser după a doua doză Nu mai este s-a observat acumulare relevantă clinic La majoritatea pacienților cu boală Crohn fistulizantă, infliximab a fost detectat în ser timp de 12 săptămâni (interval 4-28 săptămâni) după administrarea regimului.
Populația pediatrică
O „analiză farmacocinetică populațională bazată pe date obținute de la pacienții cu colită ulcerativă (N = 60), boala Crohn (N = 112), artrita reumatoidă juvenilă (N = 117) și boala Kawasaki (N = 16) cu vârsta de 2 luni până la În general, 17 ani au indicat faptul că expunerea la infliximab a fost dependentă neliniar de greutatea corporală. După administrarea a 5 mg / kg de Remicade la fiecare 8 săptămâni, expunerea mediană așteptată la infliximab la starea de echilibru (zona sub curba concentrație-timp la starea de echilibru, ASC) la copii și adolescenți cu vârsta cuprinsă între 6 și 17 ani a fost cu aproximativ 20% mai mică decât mediana a prezis expunerea la medicament la starea de echilibru la adulți. AUCss mediană la copii și adolescenți cu vârsta cuprinsă între 2 și mai puțin de 6 ani s-a prezis a fi cu aproximativ 40% mai mică decât la adulți, deși numărul pacienților care susțin această estimare este limitat.
05.3 Date preclinice de siguranță
Infliximab nu reacționează încrucișat cu TNFα la alte specii de animale decât oamenii și cimpanzeii. Prin urmare, datele preclinice convenționale de siguranță cu infliximab sunt limitate. Într-un studiu de toxicitate asupra dezvoltării șoarecilor folosind un anticorp similar care inhibă selectiv activitatea funcțională a șoarecelui TNFα nu a fost găsit, toxicitate maternă, embriotoxicitate, teratogenitate. Într-un studiu privind fertilitatea și funcția reproductivă generală, numărul șoarecilor însărcinați a fost redus după administrarea aceluiași anticorp analog. Nu se știe dacă aceste constatări s-au datorat efectelor asupra bărbaților și / sau femeilor.Într-un studiu de toxicitate cu doze repetate de 6 luni la șobolani, folosind aceiași anticorpi analogi la TNFα murin, s-au observat depozite cristaline pe capsula lentilei unora dintre șobolanii masculi tratați. Nu a fost efectuată nicio examinare oftalmologică specifică pacienților pentru a evalua relevanța acestui eveniment la om. Nu s-au efectuat studii pe termen lung pentru a evalua potențialul cancerigen al infliximabului. În studiile efectuate pe șoareci cu deficit de TNFα, s-a demonstrat că nu există o creștere a tumorilor atunci când sunt provocate cu inițiatori și / sau promotori de tumori cunoscuți.
06.0 INFORMAȚII FARMACEUTICE
06.1 Excipienți
Zaharoza
Polisorbat 80
Fosfat de sodiu monobazic
Fosfat de sodiu dibazic
06.2 Incompatibilitate
În absența studiilor de compatibilitate, acest medicament nu trebuie amestecat cu alte medicamente.
06.3 Perioada de valabilitate
Înainte de reconstituire:
3 ani la 2 ° C - 8 ° C.
Remicade poate fi păstrat la temperaturi nu mai mari de 25 ° C pentru o singură perioadă de până la 6 luni, dar nu după data de expirare inițială. Noua dată de expirare trebuie să fie scrisă pe cutie. După scoaterea din frigider, Remicade nu trebuie păstrat din nou în frigider.
După reconstituire:
Stabilitatea chimică și fizică la utilizare a soluției reconstituite a fost demonstrată timp de 24 de ore la 25 ° C. Din punct de vedere microbiologic, produsul trebuie utilizat cât mai curând posibil și, în orice caz, în termen de 3 ore de la reconstituire și diluare. . Dacă nu este utilizat imediat, timpii și condițiile de păstrare în timpul utilizării sunt responsabilitatea utilizatorului și în niciun caz nu trebuie să depășească 24 de ore la 2 ° C până la 8 ° C.
06.4 Precauții speciale pentru depozitare
A se păstra la frigider (2 ° C - 8 ° C).
Pentru condiții de depozitare de până la 25 ° C înainte de reconstituirea medicamentului, vezi pct. 6.3.
Pentru condițiile de depozitare după reconstituirea medicamentului, vezi pct. 6.3.
06.5 Natura ambalajului imediat și conținutul ambalajului
Flacon de sticlă tip I cu dop de cauciuc și sertizare din aluminiu protejat de un capac din plastic, conținând 100 mg infliximab.
Remicade este disponibil în pachete de 1, 2, 3, 4 sau 5 flacoane. Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
06.6 Instrucțiuni de utilizare și manipulare
1. Calculați doza și numărul de flacoane Remicade necesare. Fiecare flacon de Remicade conține 100 mg infliximab. Calculați volumul total necesar soluției de Remicade reconstituită.
2. În condiții aseptice, reconstituiți fiecare flacon de Remicade cu 10 ml de apă pentru preparate injectabile folosind o seringă cu un ac de calibru 21 (0,8 mm) sau mai mic. Scoateți clema de aluminiu din flacon și curățați capacul cu un tampon de bumbac înmuiat în alcool 70%. Introduceți acul seringii în flacon prin centrul dopului de cauciuc și direcționați fluxul de apă pentru injecții către peretele de sticlă al flaconului. Rotiți ușor soluția pentru a dizolva complet pulberea liofilizată. Nu agitați energic sau pentru o lungă perioadă de timp Nu agitați. În timpul reconstituirii poate apărea spumare. Lăsați soluția reconstituită să stea timp de 5 minute. Verificați dacă soluția este incoloră până la galbenă și opalescentă, soluția poate avea câteva particule translucide mici, deoarece infliximab este o proteină. soluția dacă observați particule plictisitoare, schimbări de culoare sau alte corpuri străine.
3. Diluați volumul total al dozei de soluție reconstituită Remicade la 250 ml utilizând soluție perfuzabilă de clorură de sodiu 9 mg / ml (0,9%). Nu diluați soluția reconstituită Remicade cu niciun alt diluant. Acest lucru se poate face prin extragerea unui volum de soluție perfuzabilă de clorură de sodiu 9 mg / ml (0,9%) din flaconul de sticlă de 250 ml sau punga de perfuzie egală cu volumul de Remicade reconstituit. Adăugați încet volumul total de soluție reconstituită Remicade în flaconul de 250 ml sau punga de perfuzie. Se amestecă ușor.
4. Administrați soluția de perfuzie într-un timp de perfuzie de cel puțin timpul de perfuzie recomandat (vezi pct. 4.2). Utilizați numai un set de perfuzie cu un filtru în linie steril, nepirogen, cu un nivel scăzut de legare a proteinelor (diametrul porilor de 1,2 micrometri sau mai puțin). Deoarece nu conține conservant, se recomandă începerea administrării soluției pentru perfuzie intravenoasă cât mai curând posibil și în termen de 3 ore de la reconstituire și diluare. Dacă reconstituirea și diluarea se fac în condiții aseptice, soluția de perfuzie Remicade poate fi utilizată în 24 de ore când se păstrează la 2 ° C până la 8 ° C. Soluția neutilizată nu trebuie păstrată pentru o utilizare ulterioară.
5. Nu au fost efectuate studii de compatibilitate fizică și biochimică pentru a evalua combinația Remicade cu alți agenți. Nu administrați Remicade concomitent cu alte medicamente în aceeași linie intravenoasă.
6. Înainte de administrare, inspectați vizual Remicade pentru a vă asigura că nu se observă particule sau decolorări. Dacă se observă particule opace, decolorare sau particule străine, nu utilizați.
7. Medicamentele neutilizate și deșeurile derivate din acest medicament trebuie eliminate în conformitate cu reglementările locale.
07.0 DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Janssen Biologics B.V. Einsteinweg 101
2333 CB Leiden
Olanda
08.0 NUMĂRUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU / 1/99/116/001
034528012
EU / 1/99/116/002
EU / 1/99/116/003
EU / 1/99/116/004
EU / 1/99/116/005
09.0 DATA PRIMEI AUTORIZAȚII SAU REÎNNOIREA AUTORIZAȚIEI
Data primei autorizații: 13 august 1999. Data celei mai recente reînnoiri: 02 iulie 2009.
10.0 DATA REVIZUIRII TEXTULUI
24 septembrie 2015