Ingrediente active: Denosumab
XGEVA 120 mg soluție injectabilă
Indicații De ce se utilizează Xgeva? Pentru ce este?
XGEVA conține denosumab, o proteină (anticorp monoclonal) care acționează pentru scăderea distrugerii osoase cauzate de răspândirea cancerului la nivelul osului (metastaze osoase) sau a cancerului osos cu celule gigantice.
XGEVA este utilizat la adulții cu cancer pentru a preveni complicațiile grave cauzate de metastaze osoase (de exemplu, fracturi, presiune asupra măduvei osoase sau necesitatea unei radioterapii sau a unei intervenții chirurgicale). XGEVA este, de asemenea, utilizat pentru tratamentul cancerului osos cu celule uriașe, care nu poate fi tratat prin intervenție chirurgicală sau în care intervenția chirurgicală nu este cea mai bună opțiune, la adulți și adolescenți ale căror oase au încetat să crească.
Contraindicații Când nu trebuie utilizat Xgeva
Nu utilizați XGEVA
- dacă sunteți alergic la denosumab sau la oricare dintre celelalte componente ale XGEVA.
Personalul medical nu vă va administra XGEVA dacă aveți un nivel foarte scăzut de calciu în sânge care nu a fost tratat.
Profesionistul dumneavoastră din domeniul sănătății nu vă va administra XGEVA dacă aveți răni care nu s-au vindecat în urma unei intervenții chirurgicale dentare sau orale.
Precauții pentru utilizare Ce trebuie să știți înainte să luați Xgeva
Supliment de calciu și vitamina D
Trebuie să luați suplimente de calciu și vitamina D în timp ce sunteți tratat cu XGEVA, cu excepția cazului în care nivelul de calciu din sânge este ridicat. Medicul dumneavoastră vă va discuta acest lucru cu dumneavoastră. Dacă nivelul de calciu din sânge este scăzut, medicul dumneavoastră poate decide să vă ofere suplimente de calciu înainte de a începe tratamentul cu XGEVA.
Niveluri scăzute de calciu în sânge
Spuneți imediat medicului dumneavoastră dacă aveți spasme musculare, zvâcniri sau crampe și / sau amorțeală sau furnicături la nivelul degetelor și de la picioare sau în jurul gurii și / sau convulsii, confuzie sau pierderea cunoștinței în timp ce luați XGEVA. Este posibil să aveți niveluri scăzute de calciu în sânge.
Spuneți medicului dumneavoastră dacă aveți sau ați suferit vreodată de probleme renale severe, insuficiență renală sau dacă ați fost supus dializei, deoarece poate crește riscul de a avea niveluri scăzute de calciu din sânge, mai ales dacă nu luați suplimente de calciu.
Probleme cu gura, dinții sau maxilarul
Un efect secundar numit osteonecroză a maxilarului (degenerescență osoasă severă a maxilarului) a fost raportat frecvent (poate afecta până la 1 din 10 persoane) la pacienții cărora li se administrează injecții cu XGEVA pentru afecțiuni legate de cancer.
Osteonecroza maxilarului poate apărea și după oprirea tratamentului.
Este important să încercați să preveniți dezvoltarea osteonecrozei maxilarului, deoarece este o afecțiune dureroasă care poate fi dificil de tratat.Pentru a reduce riscul de apariție a osteonecrozei maxilarului, trebuie să luați anumite precauții.
Înainte de a primi tratament, spuneți medicului / asistentei (medicului) dacă aveți probleme cu gura sau dinții. Medicul dumneavoastră ar trebui să întârzie începerea tratamentului dacă aveți răni în gură care nu s-au vindecat în urma procedurilor dentare sau a intervențiilor chirurgicale orale.
În timpul tratamentului este necesar să se mențină o igienă orală bună și să se efectueze controale dentare periodice. Dacă purtați proteze, trebuie să vă asigurați că sunt introduse corect.
Dacă urmează un tratament dentar sau intenționați să efectuați o intervenție chirurgicală dentară (de exemplu, extracții dentare), vă rugăm să informați medicul dumneavoastră de tratament dentar și să îl informați pe dentist că sunteți tratat cu XGEVA.
Contactați imediat medicul și medicul dentist dacă observați probleme cu gura sau dinții, cum ar fi oscilația dinților, durere sau umflături, sau dacă rănile sau descărcarea gurii nu se vindecă, deoarece acestea ar putea fi semne de osteonecroză a mandibulei / maxilarului .
Pacienții supuși chimioterapiei și / sau radioterapiei, care iau steroizi sau medicamente anti-angiogene (utilizate pentru tratarea cancerului), care suferă o intervenție chirurgicală dentară, care nu primesc îngrijiri dentare de rutină sau care suferă de gingii, sunt fumători, pot avea un risc mai mare de a dezvolta osteonecroză a maxilarului .
Fracturi neobișnuite ale osului coapsei
Unii oameni au dezvoltat fracturi neobișnuite la femur în timp ce erau tratați cu XGEVA. Adresați-vă medicului dumneavoastră dacă aveți dureri noi sau neobișnuite la nivelul șoldului, inghinei sau coapsei.
Copii și adolescenți
XGEVA nu este recomandat copiilor și adolescenților cu vârsta sub 18 ani, cu excepția adolescenților cu tumoră osoasă cu celule uriașe ale căror oase au încetat să crească. Utilizarea XGEVA la copii și adolescenți cu alte tumori care au invadat osul nu a fost studiată.
Interacțiuni Ce medicamente sau alimente pot schimba efectul Xgeva
Spuneți medicului dumneavoastră sau farmacistului dacă luați sau ați luat recent sau ați putea lua orice alte medicamente, inclusiv medicamente eliberate fără prescripție medicală. În special, este important să spuneți medicului dumneavoastră dacă luați
- un alt medicament care conține denosumab
- un bifosfonat.
Nu trebuie să luați XGEVA împreună cu alte medicamente care conțin denosumab sau care conțin bifosfonat
Avertismente Este important să știm că:
Sarcina și alăptarea
XGEVA nu a fost studiat la femeile gravide. Este important să spuneți medicului dumneavoastră dacă sunteți gravidă, suspectați sau intenționați să rămâneți gravidă. Utilizarea XGEVA nu este recomandată dacă sunteți gravidă.Femeile aflate la vârsta fertilă trebuie să utilizeze metode contraceptive eficiente în timp ce luați XGEVA și timp de cel puțin 5 luni după întreruperea tratamentului cu XGEVA.
Dacă rămâneți gravidă în timpul tratamentului cu XGEVA sau la mai puțin de 5 luni de la întreruperea tratamentului cu XGEVA, vă rugăm să informați medicul dumneavoastră. Este încurajată să se înscrie în Programul de supraveghere a sarcinii de la Amgen. Detaliile reprezentantului local Amgen sunt date în secțiunea 6 a acestui prospect.
Nu se știe dacă XGEVA este excretat în laptele uman. Este important să spuneți medicului dumneavoastră dacă alăptați sau intenționați să alăptați. Medicul dumneavoastră vă va ajuta apoi să decideți dacă întrerupeți alăptarea sau să încetați să luați XGEVA, ținând cont de beneficiile alăptării pentru copil și de beneficiul administrării XGEVA pentru mamă.
Dacă alăptați în timp ce luați XGEVA, vă rugăm să spuneți medicului dumneavoastră. Este încurajată să se înscrie în Programul de supraveghere a lactației Amgen.Detaliile reprezentantului local Amgen sunt date în secțiunea 6 a acestui prospect.
Adresați-vă medicului dumneavoastră sau farmacistului pentru recomandări înainte de a utiliza acest medicament.
Conducerea vehiculelor și utilizarea utilajelor
XGEVA nu are nicio influență sau are o influență neglijabilă asupra capacității de a conduce vehicule sau de a folosi utilaje.
XGEVA conține sorbitol
Dacă medicul dumneavoastră v-a spus că aveți o „intoleranță la unele zaharuri, contactați medicul înainte de a lua acest medicament, deoarece conține sorbitol (E420).
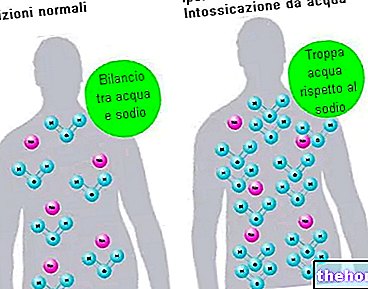
XGEVA conține sodiu
Acest medicament conține mai puțin de 1 mmol sodiu (23 mg) la 120 mg, adică în esență „fără sodiu”.
Doză, metodă și timp de administrare Cum se utilizează Xgeva: Doze
Doza recomandată de XGEVA este de 120 mg administrată o dată la 4 săptămâni, sub formă de injecție unică sub piele (subcutanată). XGEVA va fi injectat la nivelul coapsei, abdomenului sau brațului superior. Dacă sunteți tratat pentru cancerul osos cu celule uriașe, veți primi o doză suplimentară la 1 săptămână și 2 săptămâni după prima doză.
XGEVA trebuie administrat sub responsabilitatea unui cadru medical.
Nu agitați excesiv.
De asemenea, trebuie să luați suplimente de calciu și vitamina D în timpul tratamentului cu XGEVA. Medicul dumneavoastră vă va discuta acest lucru cu dumneavoastră.
Dacă aveți orice întrebări suplimentare cu privire la acest medicament, adresați-vă medicului dumneavoastră, farmacistului sau asistentei medicale.
Efecte secundare Care sunt efectele secundare ale Xgeva
Ca toate medicamentele, acest medicament poate provoca reacții adverse, deși nu apar la toate persoanele.
Spuneți imediat medicului dumneavoastră dacă observați oricare dintre aceste simptome în timp ce luați XGEVA:
- zvâcniri, zvâcniri, crampe musculare, amorțeală sau furnicături la nivelul degetelor și de la picioare sau în jurul gurii și / sau convulsii, confuzie sau pierderea cunoștinței. Aceste semne ar putea indica niveluri scăzute de calciu din sânge. Nivelurile scăzute de calciu din sânge pot duce, de asemenea, la o schimbare a ritmului cardiac numită prelungire QT, care se observă în electrocardiografie (ECG).
Spuneți imediat medicului și medicului dentist dacă aveți oricare dintre aceste simptome în timpul tratamentului cu XGEVA sau după oprirea tratamentului cu XGEVA:
- durere la nivelul gurii și / sau maxilarului, umflături sau răni nevindecătoare la nivelul gurii sau maxilarului, descărcare, amorțeală sau senzație de greutate la nivelul maxilarului sau balansarea unui dinte, deoarece aceste semne ar putea indica o degenerare osoasă severă a maxilarului ( osteonecroză).
Reacții adverse foarte frecvente (pot afecta mai mult de 1 din 10 persoane):
- dureri la nivelul oaselor, articulațiilor și / sau mușchilor uneori severe,
- respirație șuierătoare (dispnee),
- diaree.
Reacții adverse frecvente (pot afecta până la 1 din 10 persoane):
- niveluri scăzute de calciu în sânge (hipocalcemie),
- niveluri scăzute de fosfat în sânge (hipofosfatemie),
- durere persistentă și / sau non-vindecare a rănilor la nivelul gurii sau maxilarului (osteonecroză a maxilarului),
- extracție dentară,
- transpirație excesivă.
Reacții adverse rare (pot afecta până la 1 din 1000 de persoane):
- reacții alergice (de exemplu respirație șuierătoare sau dificultăți de respirație; umflarea feței, buzelor, limbii, gâtului sau a altor părți ale corpului; erupții cutanate, mâncărime sau urticarie pe piele). În cazuri rare, reacțiile alergice pot fi severe.
- o durere nouă sau neobișnuită la nivelul șoldului, inghinei sau coapsei (acesta ar putea fi un semn precoce al unei posibile fracturi osoase a coapsei).
Raportarea efectelor secundare
Dacă manifestați orice reacție adversă, discutați cu medicul sau asistenta medicală.Acestea includ orice posibile reacții adverse nemenționate în acest prospect.Puteți raporta, de asemenea, reacțiile adverse direct prin intermediul sistemului național de raportare enumerat în Anexa V. Raportând reacțiile adverse puteți ajuta furnizați mai multe informații despre siguranța acestui medicament.
Expirare și reținere
Nu lăsați acest medicament la vederea și îndemâna copiilor.
Nu utilizați acest medicament după data de expirare înscrisă pe etichetă și cutie după EXP. Data de expirare se referă la ultima zi a lunii respective.
A se păstra la frigider (2 ° C - 8 ° C).
Nu înghețați.
A se păstra în ambalajul original pentru a proteja medicamentul de lumină.
Flaconul poate fi lăsat afară din frigider pentru a atinge temperatura camerei (până la 25 ° C) înainte de injecție, ceea ce va face injecția mai confortabilă. Odată ce flaconul a atins temperatura camerei (până la 25 ° C), acesta trebuie utilizat în termen de 30 de zile.
Nu aruncați niciun medicament prin apele uzate sau deșeurile menajere. Întrebați farmacistul cum să aruncați medicamentele pe care nu le mai utilizați. Acest lucru va ajuta la protejarea mediului.
Termen limită "> Alte informații
Ce conține XGEVA
- Ingredientul activ este denosumab. Fiecare flacon conține 120 mg în 1,7 ml de soluție (corespunzător la 70 mg / ml).
- Celelalte componente sunt acid acetic glacial, hidroxid de sodiu, sorbitol (E420) și apă pentru preparate injectabile.
Descrierea aspectului XGEVA și conținutul ambalajului
XGEVA este o soluție injectabilă într-un flacon.
Fiecare pachet conține unul, trei sau patru flacoane.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
XGEVA este o soluție limpede, incoloră până la ușor galbenă. Poate conține urme de particule limpezi până la albe.
Prospect sursă: AIFA (Agenția italiană pentru medicamente). Conținut publicat în ianuarie 2016. Este posibil ca informațiile prezente să nu fie actualizate.
Pentru a avea acces la cea mai actualizată versiune, este recomandabil să accesați site-ul web AIFA (Agenția italiană pentru medicamente). Declinare de responsabilitate și informații utile.
01.0 DENUMIREA MEDICAMENTULUI -
XGEVA 120 MG Soluție pentru injectare
▼ Medicament supus unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă a noilor informații de siguranță. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată. Vezi pct. 4.8 pentru informații despre cum să raportezi reacțiile adverse.
02.0 COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ -
Fiecare flacon conține 120 mg denosumab în 1,7 ml soluție (70 mg / ml).
Denosumab este un anticorp monoclonal tip IgG2 uman produs într-o linie celulară de mamifere (CHO) prin tehnologia ADN-ului recombinant.
Excipienți cu efect cunoscut
Fiecare 1,7 ml soluție conține 78 mg sorbitol (E420).
Pentru lista completă a excipienților, vezi secțiunea 6.1.
03.0 FORMA FARMACEUTICĂ -
Soluție injectabilă (injecție).
Soluție limpede, incoloră până la ușor galbenă, care poate conține urme de particule de proteine translucide până la albe.
04.0 INFORMAȚII CLINICE -
04.1 Indicații terapeutice -
Prevenirea evenimentelor legate de schelet (fracturi patologice, radioterapie osoasă, compresie a măduvei spinării sau intervenții chirurgicale osoase) la adulți cu metastaze osoase din tumori solide.
Tratamentul adulților și adolescenților maturi din punct de vedere scheletic cu tumoră osoasă nerezecabilă cu os sau pentru care rezecția chirurgicală ar putea provoca morbiditate severă.
04.2 Doze și mod de administrare -
XGEVA trebuie administrat sub responsabilitatea unui cadru medical.
Dozare
Suplimentarea a cel puțin 500 mg de calciu și 400 UI de vitamina D pe zi este necesară la toți pacienții, cu excepția hipercalcemiei (vezi pct. 4.4).
Pacienții tratați cu XGEVA trebuie să primească prospectul și fișa de reamintire a pacientului.
Prevenirea evenimentelor legate de schelet la adulții cu metastaze osoase din tumori solide
Doza recomandată este de 120 mg administrată sub formă de injecție subcutanată unică o dată la 4 săptămâni la nivelul coapsei, abdomenului sau brațului superior.
Tumora cu celule uriașe a osului
Doza recomandată de XGEVA este de 120 mg, administrată sub formă de injecție subcutanată unică, o dată la 4 săptămâni la nivelul coapsei, abdomenului sau brațului superior, cu doze suplimentare de 120 mg în zilele 8 și 15 de tratament în prima lună de tratament.
Pacienții din studiul de fază II care au suferit rezecția completă a tumorii osoase cu celule gigantice au primit încă 6 luni de tratament după operație conform protocolului de studiu.
Pacienții cu cancer osos cu celule gigantice trebuie evaluați la intervale regulate pentru a determina dacă beneficiază în continuare de tratament. La pacienții a căror boală este controlată de XGEVA, efectul opririi sau întreruperii tratamentului nu va fi fost evaluat, cu toate acestea, există date limitate în aceste pacienții nu indică un efect de revenire la întreruperea tratamentului.
Pacienți cu insuficiență renală
Nu este necesară ajustarea dozei la pacienții cu insuficiență renală (vezi pct. 4.4 pentru recomandări privind monitorizarea concentrațiilor de calciu, 4.8 și 5.2).
Pacienți cu insuficiență hepatică
Siguranța și eficacitatea denosumab nu au fost studiate la pacienții cu insuficiență hepatică (vezi pct. 5.2).
Pacienți vârstnici (vârsta ≥ 65 ani)
Nu este necesară ajustarea dozei la pacienții vârstnici (vezi pct. 5.2).
Populația pediatrică
Siguranța și eficacitatea XGEVA nu au fost stabilite la copii și adolescenți (vârstă
XGEVA nu este recomandat la copii și adolescenți (vârstă
Tratamentul adolescenților maturi din punct de vedere scheletic cu tumoră osoasă nerezecabilă cu os sau la care rezecția chirurgicală ar putea provoca morbiditate severă: posologia este aceeași cu cea a adulților.
În studiile la animale, inhibarea ligandului RANK / RANK (RANKL) a fost asociată cu inhibarea creșterii osoase și a eșecului erupției și aceste modificări au fost parțial reversibile după întreruperea inhibării RANKL (vezi paragraful 5.3).
Mod de administrare
Pentru utilizare subcutanată.
Pentru instrucțiuni de utilizare, manipulare și eliminare, consultați secțiunea 6.6.
04.3 Contraindicații -
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
Hipocalcemie severă, netratată (vezi pct. 4.4).
Leziuni care nu au fost vindecate prin intervenții chirurgicale dentare sau orale.
04.4 Avertismente speciale și precauții adecvate pentru utilizare -
Supliment de calciu și vitamina D
Este important ca toți pacienții să primească un aport adecvat de calciu și vitamina D, cu excepția cazului de hipercalcemie (vezi pct. 4.2).
Hipocalcemie
Hipocalcemia preexistentă trebuie corectată înainte de inițierea tratamentului cu XGEVA.
Hipocalcemia poate apărea în orice moment în timpul tratamentului cu XGEVA. Monitorizarea nivelurilor de calciu trebuie făcută înainte de doza inițială de XGEVA, în termen de două săptămâni de la administrarea dozei inițiale, în caz de suspiciune de simptome de hipocalcemie (vezi pct. 4.8 pentru lista simptomelor) . Monitorizarea suplimentară a nivelurilor de calciu trebuie luată în considerare în timpul tratamentului la pacienții cu factori de risc pentru hipocalcemie sau după cum se indică altfel în funcție de starea clinică a pacientului.
Pacienții trebuie încurajați să raporteze simptome care indică hipocalcemie. Dacă se produce hipocalcemie în timpul administrării XGEVA, sunt necesare suplimente suplimentare de calciu și monitorizare suplimentară.
În timpul utilizării după punerea pe piață, a fost raportată hipocalcemie simptomatică severă (inclusiv cazuri letale) (vezi pct. 4.8), majoritatea cazurilor apărând în primele săptămâni de la inițierea tratamentului, dar pot apărea mai târziu.
Insuficiență renală
Pacienții cu insuficiență renală severă (dializa cu clearance-ul creatininei prezintă un risc crescut de a dezvolta hipocalcemie. Riscul de a dezvolta hipocalcemie și, prin urmare, creșterea nivelurilor hormonilor paratiroidieni crește odată cu creșterea gradului de insuficiență renală. Monitorizarea regulată a nivelurilor bolilor renale. Calciu este deosebit de important în acești pacienți.
Osteonecroza maxilarului (ONJ)
ONJ a fost raportată frecvent la pacienții cărora li s-a administrat XGEVA (vezi pct. 4.8).
Inițierea tratamentului / tratamentul nou trebuie amânat la pacienții cu leziuni ale țesuturilor moi, nevindecate, deschise la nivelul gurii.
Următorii factori de risc trebuie luați în considerare la evaluarea riscului unui pacient de a dezvolta ONJ:
• potența medicamentului care inhibă resorbția osoasă (riscul este mai mare cu medicamente mai puternice), calea de administrare (riscul este mai mare cu administrarea parenterală) și doza cumulativă a terapiei de resorbție osoasă.
• tumori, afecțiuni comorbide (de exemplu anemie, coagulopatii, infecție), fumat.
• terapii concomitente: corticosteroizi, chimioterapie, inhibitori ai angiogenezei, radioterapie a regiunii capului și gâtului.
• igienă orală deficitară, boală parodontală, proteze dentare introduse incorect, boală dentară preexistentă, proceduri dentare invazive (de exemplu, extracții dentare).
Toți pacienții trebuie încurajați să mențină o igienă orală bună, să facă controale dentare periodice și să raporteze imediat orice simptome orale, cum ar fi mobilitatea dinților, durerea sau umflarea sau nevindecarea rănilor bucale sau prezența secrețiilor în timpul tratamentului. . În timpul tratamentului, procedurile dentare invazive trebuie efectuate numai după o analiză atentă și trebuie evitate în imediata apropiere a administrării XGEVA.
Managementul pacienților care dezvoltă ONJ trebuie făcut în strânsă colaborare între medicul curant și un dentist sau chirurg maxilo-facial cu experiență în tratamentul ONJ. Întreruperea temporară a tratamentului cu XGEVA trebuie luată în considerare până la rezolvarea afecțiunii și, acolo unde este posibil, pentru a atenua factorii de risc care au contribuit la apariția acesteia.
Fracturi atipice ale femurului
Au fost raportate cazuri de fracturi femurale atipice la pacienții tratați cu XGEVA (vezi pct. 4.8). Fracturile femurale atipice pot apărea cu traume minime sau deloc în regiunile subtrochanterice și diafizare ale femurului. Aceste evenimente sunt caracterizate de descoperiri radiografice specifice. Au fost raportate, de asemenea, fracturi femurale atipice la pacienții cu unele afecțiuni comorbide (de exemplu, deficit de vitamina D, artrită reumatoidă, hipofosfatazie) și în utilizarea anumitor medicamente (de exemplu, bifosfonați, glucocorticoizi, inhibitori ai pompei de protoni). Aceste evenimente au apărut și în absența terapiei antiresorptive. Fracturile similare, raportate în asociere cu utilizarea bifosfonaților, sunt adesea bilaterale; prin urmare, femurul contralateral ar trebui evaluat la pacienții tratați cu denosumab care au suferit o fractură a arborelui femural. La pacienții cu suspiciune de fractură femurală atipică, trebuie luată în considerare încetarea XGEVA, în așteptarea evaluării pacientului pe baza unei analize individuale a raportului beneficiu / risc.În timpul tratamentului cu XGEVA, pacienții trebuie sfătuiți să raporteze dureri noi sau neobișnuite la nivelul coapsei, șoldului sau inghinei. Pacienții care prezintă astfel de simptome trebuie evaluați pentru o fractură femurală incompletă.
Pacienți cu un sistem osos în creștere
XGEVA nu este recomandat la pacienții cu sisteme scheletice în creștere (vezi pct. 4.2). S-a raportat hipercalcemie semnificativă clinic la pacienții cu sisteme scheletice în creștere, tratați cu XGEVA după săptămâni până la luni de întrerupere a tratamentului.
Alții
Pacienții care sunt tratați cu XGEVA nu trebuie tratați concomitent cu alte medicamente care conțin denosumab (pentru indicații de osteoporoză).
Pacienții care sunt tratați cu XGEVA nu trebuie tratați concomitent cu bifosfonați.
Degenerarea tumorii cu celule gigantice a bolii maligne sau progresia metastatică a bolii sunt evenimente rare și reprezintă un risc cunoscut la pacienții cu tumoră osoasă cu celule gigantice. Pacienții trebuie monitorizați pentru semne radiologice de malignitate, radiotransparență nouă sau osteoliză. Datele clinice disponibile nu sugerează un risc crescut de malignitate la pacienții cu tumoră osoasă cu celule gigant tratate cu XGEVA.
Avertismente pentru excipienți
XGEVA conține sorbitol. Pacienții cu afecțiuni ereditare rare de intoleranță la fructoză nu trebuie să ia XGEVA.
Acest medicament conține mai puțin de 1 mmol sodiu (23 mg) la 120 mg, adică în esență „fără sodiu”.
04.5 Interacțiuni cu alte medicamente și alte forme de interacțiune -
Nu au fost efectuate studii de interacțiune.
În studiile clinice, XGEVA a fost administrat în asociere cu tratamente antitumorale standard și la pacienții tratați anterior cu bifosfonați. Nu au existat modificări relevante din punct de vedere clinic ale concentrației plasmatice și farmacodinamicii denosumab (N-telopeptidă ajustată la creatinină urinară, uNTx / Cr) datorită terapiei hormonale și / sau chimioterapiei concomitente sau administrării intravenoase anterioare de bifosfonați.
04.6 Sarcina și alăptarea -
Sarcina
Nu sunt disponibile date adecvate cu privire la utilizarea XGEVA la femeile gravide. Toxicitatea asupra funcției de reproducere a fost demonstrată într-un studiu efectuat pe maimuțe cynomolgus cu administrare de denosumab în timpul sarcinii cu ASC de 12 ori mai mare decât doza umană (vezi pct. 5.3.).
Utilizarea XGEVA nu este recomandată femeilor gravide și femeilor aflate la vârsta fertilă care nu utilizează contraceptive extrem de eficiente. Femeile trebuie sfătuite să evite să rămână gravide în timpul tratamentului cu XGEVA și timp de cel puțin 5 luni după tratament. Tratament. XGEVA este probabil să fie mai mare în timpul celui de-al doilea și al treilea trimestru de sarcină, deoarece anticorpii monoclonali sunt transportați peste placentă într-un mod liniar pe măsură ce sarcina progresează, cea mai mare cantitate fiind transferată în timpul celui de-al treilea trimestru de sarcină.
Timp de hrănire
Nu se știe dacă denosumab este excretat în laptele matern uman. Studiile efectuate la șoareci knockout sugerează că absența RANKL în timpul sarcinii ar putea interfera cu maturarea glandei mamare, ducând la lactație afectată după naștere (vezi secțiunea 5.3). Trebuie luată o decizie dacă să se abțină de la alăptare sau de la terapia XGEVA, luând în considerare beneficiul alăptării pentru nou-născut / sugar și beneficiul terapiei XGEVA pentru femeie.
Fertilitate
Nu există date privind efectele denosumab asupra fertilității umane. Studiile la animale nu indică efecte dăunătoare directe sau indirecte asupra fertilității (vezi pct. 5.3).
04.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje -
XGEVA nu are nicio influență sau are o influență neglijabilă asupra capacității de a conduce vehicule și de a folosi utilaje.
04.8 Efecte nedorite -
Rezumatul profilului de siguranță
Profilul general de siguranță este consistent în toate indicațiile aprobate.
Hipocalcemia a fost raportată frecvent după administrarea XGEVA, în special în primele 2 săptămâni. Hipocalcemia poate fi severă și simptomatică (vezi pct. 4.8 - descrierea reacțiilor adverse selectate). Scăderile concentrațiilor serice de calciu sunt de obicei gestionate în mod adecvat cu suplimentarea cu calciu și vitamina D. Cele mai frecvente reacții adverse cu XGEVA sunt durerile musculo-scheletice.
Siguranța XGEVA a fost evaluată pe:
• 5.931 de pacienți cu cancer avansat care implică os în studii clinice active controlate care evaluează eficacitatea și siguranța XGEVA față de acidul zoledronic în prevenirea evenimentelor legate de schelet.
• 523 pacienți cu tumoră osoasă cu celule gigant într-un studiu clinic cu un singur braț pentru a evalua eficacitatea și siguranța XGEVA.
Reacțiile adverse identificate în aceste studii clinice și în contextul post-comercializare sunt prezentate în Tabelul 1.
Tabelul reacțiilor adverse
Pentru clasificarea reacțiilor adverse pe baza ratelor de incidență în trei studii clinice de fază III și două faze II, a fost utilizată următoarea convenție (vezi tabelul 1): foarte frecvente (≥ 1/10), frecvente (≥ 1/100,
Tabelul 1: Reacții adverse raportate la pacienții cu cancer avansat care implică cancer osos sau cu celule uriașe
¹ A se vedea secțiunea Descrierea reacțiilor adverse selectate
² A se vedea secțiunea Alte populații speciale
Descrierea reacțiilor adverse selectate
Hipocalcemie
În trei studii clinice de fază III controlate activ la pacienți cu cancer avansat care implică os, s-a raportat hipocalcemie la 9,6% dintre pacienții tratați cu XGEVA și 5,0% dintre pacienții tratați cu acid zoledronic.
O scădere de gradul 3 a concentrațiilor plasmatice de calciu a fost detectată la 2,5% dintre pacienții tratați cu XGEVA și la 1,2% dintre pacienții tratați cu acid zoledronic. dintre pacienții tratați cu acid zoledronic (vezi pct. 4.4).
În două studii clinice de fază II cu un singur braț la pacienți cu tumoră osoasă cu celule gigant, s-a raportat hipocalcemie la 5,7% dintre pacienți. Niciunul dintre evenimentele adverse nu a fost considerat grav.
În timpul utilizării după punerea pe piață, a fost raportată hipocalcemie simptomatică severă (inclusiv cazuri letale), majoritatea cazurilor apărând în primele câteva săptămâni de la inițierea tratamentului. Exemple de manifestări clinice de hipocalcemie simptomatică severă au inclus prelungirea intervalului QT, tetanie, convulsii și modificarea stării psihice (inclusiv comă) (vezi pct. 4.4).
Osteonecroza maxilarului (ONJ)
În studiile clinice, incidența ONJ a fost mai mare cu o durată mai lungă de expunere; ONJ a fost, de asemenea, diagnosticată după încheierea tratamentului cu XGEVA, majoritatea cazurilor apărând în decurs de 5 luni de la ultima doză. Pacienții cu antecedente de ONJ sau osteomielită a mandibulei / maxilarului, cu inflamație dentară activă sau mandibulară / maxilară care necesită intervenție chirurgicală, rezultatul unei intervenții chirurgicale dentare / orale nerezolvate sau pacienții pentru care au fost planificate proceduri dentare invazive, au fost excluși din studiile clinice.
În fazele de tratament primare din trei studii clinice controlate activ de fază III la pacienți cu cancer avansat care implică os, ONJ a fost confirmată la 1,8% dintre pacienții tratați cu XGEVA (expunere mediană de 12, 0 luni; interval 0,1 - 40,5) și la 1,3 % dintre pacienții tratați cu acid zoledronic. Caracteristicile clinice ale acestor cazuri au fost similare între grupurile de tratament. Dintre pacienții cu ONJ confirmat, majoritatea (81% în ambele grupuri de tratament) au avut antecedente de extracții dentare, igienă orală slabă și / sau utilizare de aparate dentare Majoritatea subiecților au primit sau au primit chimioterapie.
Studiile clinice la pacienții cu cancer de sân sau de prostată au inclus o fază de prelungire a tratamentului cu XGEVA (expunere globală mediană de 14,9 luni; interval 0,1 - 67,2). ONJ a fost confirmat la 6,9% dintre pacienții cu cancer de sân și cancer de prostată în timpul fazei de prelungire a tratamentului.
Incidența globală confirmată a ONJ, ajustată pentru pacient-an, a fost de 1,1% în primul an de tratament, 3,7% în al doilea an și 4,6% în anii următori. 4 - 53).
În două studii clinice de fază II cu un singur braț la pacienți cu tumoră osoasă cu celule gigantice, ONJ a apărut la 2,3% (12 din 523) dintre pacienții tratați cu XGEVA (expunere globală mediană de 20,3 luni; interval: 0-83,4). Incidența ONJ, ajustată pentru anul pacientului, a fost de 0,2% în primul an de tratament și de 1,7% în al doilea an. Timpul mediu până la apariția ONJ a fost de 19,4 luni (interval: 11-40). Pe baza duratei expunerii, nu există date suficiente pentru pacienții cu GCTB pentru a evalua riscul de ONJ peste 2 ani.
Într-un studiu clinic de fază III, la pacienții cu cancer de prostată nemetastatic (o populație de pacienți pentru care XGEVA nu este indicat), cu o „expunere mai lungă la tratament (până la 7 ani), incidența ONJ confirmată, corectată pe pacient- anul a fost de 1,1% în primul an de tratament, 3,0% în al doilea an și 7,1% în anii următori.
Reacții de hipersensibilitate la medicament
Au fost raportate evenimente de hipersensibilitate, inclusiv reacții anafilactice rare, la pacienții cărora li s-a administrat XGEVA în timpul utilizării după punerea pe piață.
Fracturi atipice ale femurului
În programul de dezvoltare clinică, fracturile atipice femurale au fost raportate rar la pacienții tratați cu denosumab (vezi pct. 4.4).
Dureri musculo-scheletice
În contextul post-comercializare, durerea musculo-scheletică, inclusiv cazuri severe, a fost raportată la pacienții tratați cu XGEVA. În studiile clinice, durerea musculo-scheletică a fost foarte frecventă atât în grupurile de tratament cu denosumab, cât și cu acidul zoledronic. Durerile musculo-scheletice care au condus la întreruperea tratamentului au fost mai puțin frecvente.
Populația pediatrică
XGEVA a fost studiat într-un studiu clinic deschis care a înscris 18 adolescenți maturi din punct de vedere scheletic cu tumori osoase cu celule gigantice. Pe baza acestor date limitate, profilul evenimentelor adverse pare a fi similar cu cel la adulți.
Alte populații speciale
Insuficiență renală
Într-un studiu clinic efectuat la pacienți cu insuficiență renală severă (suplimentarea cu calciu a clearance-ului creatininei. Riscul de a dezvolta hipocalcemie în timpul tratamentului cu XGEVA este mai mare pe măsură ce crește gradul de insuficiență renală. Într-un studiu clinic la pacienți necanceri. În stadiu avansat, 19 % dintre pacienții cu insuficiență renală severă (clearance-ul creatininei
Creșteri ulterioare ale nivelurilor hormonilor paratiroidieni au fost observate și la pacienții cu insuficiență renală severă sau care primesc dializă tratați cu XGEVA. Monitorizarea nivelurilor de calciu și suplimentarea adecvată cu calciu și vitamina D sunt deosebit de importante la pacienții cu insuficiență renală (vezi pct. 4.4).
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate care apar după autorizarea medicamentului este importantă, deoarece permite monitorizarea continuă a raportului beneficiu / risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacții adverse suspectate prin intermediul sistemului național de raportare (Agenția italiană pentru medicamente - Site-ul web: http // www.agenziafarmaco.gov.it / it / manageri).
04.9 Supradozaj -
Nu au fost raportate cazuri de supradozaj în studiile clinice. În studiile clinice, XGEVA a fost administrat la doze de până la 180 mg la fiecare 4 săptămâni și 120 mg pe săptămână timp de 3 săptămâni.
05.0 PROPRIETĂȚI FARMACOLOGICE -
05.1 "Proprietăți farmacodinamice -
Grupa farmacoterapeutică: Medicamente pentru tratamentul bolilor osoase - Alte medicamente care afectează structura osoasă și mineralizarea, codul ATC: M05BX04
Mecanism de acțiune
RANKL este o proteină și se prezintă sub formă transmembranară sau solubilă. RANKL este esențial pentru formarea, funcționarea și supraviețuirea osteoclastelor, singurul tip celular responsabil de resorbția osoasă. Activitatea osteoclastică crescută, stimulată de RANKL, este un mediator cheie al distrugerii osoase în bolile osoase metastatice și în mielomul multiplu. Denosumab este un anticorp monoclonal uman (IgG2) care vizează și leagă RANKL cu afinitate și specificitate ridicate, prevenind apariția interacțiunii RANKL / RANK, reducând astfel numărul și funcția osteoclastelor, rezultând o resorbție osoasă scăzută și distrugerea osoasă indusă de cancer .
Tumorile celulare gigantice ale osului se caracterizează prin celule stromale neoplazice care exprimă ligandul RANK și celule gigantice asemănătoare osteoclastelor care exprimă RANK. La pacienții cu cancer osos cu celule gigant, denosumab se leagă de ligandul RANK, reducând semnificativ sau eliminând osteoclastul celule gigantice. În consecință, osteoliza este redusă și stroma proliferativă a tumorii este înlocuită de os nou cu o structură densă, neproliferativă, diferențiată.
Efecte farmacodinamice
În studiile clinice de fază II la pacienți cu cancer în stadiu avansat care implică administrarea subcutanată (sc) de XGEVA la fiecare 4 săptămâni sau la fiecare 12 săptămâni, a rezultat o reducere rapidă a markerilor de resorbție osoasă (uNTx / Cr, ser CTx), cu scăderi mediane de aproximativ 80% pentru uNTx / Cr în decurs de o săptămână, indiferent de terapia anterioară cu bifosfonați sau de nivelul inițial de uNTx / Cr. În studiile clinice de fază III, reducerile medii de aproximativ 80% în uNTx / Cr au fost menținute după 3 luni de tratament la 2.075 pacienți cu cancer avansat tratați cu XGEVA și tratament naif până la IV cu bifosfonați.
Imunogenitate
În studiile clinice, nu au fost observați anticorpi neutralizanți direcționați către XGEVA. Pe baza rezultatelor unei imunotestări sensibile, mai puțin de 1% dintre pacienții tratați cu denosumab timp de până la 3 ani au dat rezultate pozitive pentru anticorpi ne-neutralizanți, fără dovezi ale profilului de răspuns farmacocinetic, toxicologic sau clinic modificat.
Eficacitate clinică la pacienții cu metastaze osoase din tumori solide
Eficacitatea și siguranța XGEVA 120 mg s.c., administrată la fiecare 4 săptămâni sau acid zoledronic 4 mg i.v. (cu ajustarea dozei pentru funcția renală redusă), administrate la fiecare 4 săptămâni, au fost comparate în trei studii randomizate, dublu-orb, controlate activ la pacienți naivi cu tratament cu bifosfonat IV și cu cancere avansate cu afectare osoasă: pacienți adulți cu cancer de sân ( studiul 1), alte tumori solide sau mielom multiplu (studiu 2) și cancer de prostată rezistent la castrare (studiu 3). activ dentar sau mandibular / maxilar care necesită o intervenție chirurgicală orală, o afecțiune dentară / orală nerezolvată după o intervenție chirurgicală sau pacienți programați pentru stomatologie invazivă procedurile nu au fost eligibile pentru înscrierea la aceste studii. Obiectivele primare și secundare au evaluat apariția unuia sau mai multor evenimente legate de schelet (SRE) .În studiile care demonstrează superioritatea XGEVA față de acidul zoledronic, pacienților li s-a oferit o fază de extensie. A tratamentului pre-specificat, deschis cu XGEVA timp de 2 ani .
XGEVA a redus riscul apariției SRE și a apariției SRE multiple (prima și ulterioare) la pacienții cu metastaze osoase ale tumorilor solide (vezi Tabelul 2).
Tabelul 2: Rezultatele eficacității la pacienții cu cancer avansat care implică os
NR = nu a fost atins; NA = nu este disponibil; HCM = hipercalcemie malignă; SMR = rata morbidității scheletice; HR = raportul de pericol; RRR = reducerea relativă a riscului † Pentru studiile 1, 2 și 3, sunt prezentate valorile p ajustate (puncte finale: primul SRE și primul și următoarele SRE); * Include toate evenimentele scheletice de-a lungul timpului; sunt luate în considerare numai evenimentele care au avut loc ≥ 21 de zile după evenimentul anterior.
** Inclusiv NSCLC, cancer renal, cancer colorectal, cancer pulmonar cu celule mici, cancer de vezică urinară, cancer de cap și gât, cancer gastrointestinal / genito-urinar și alte tipuri de cancer, cu excepția cancerului de sân și de prostată
Progresia bolii și supraviețuirea generală
Progresia bolii a fost similară între XGEVA și acid zoledronic în toate cele trei studii și în analiza combinată pre-specificată a tuturor celor trei studii.
În toate cele trei studii, supraviețuirea generală între XGEVA și acid zoledronic a fost echilibrată la pacienții cu cancer avansat care implică pacienți cu cancer osos: sân (raport de risc și 95% CI: 0,95 [0,81-1,11]), pacienți cu cancer de prostată (raport de risc și 95 % CI: 1,03 [0,91-1,17]) și pacienți cu alte tumori solide sau mielom multiplu (raport de pericol și 95% CI: 0,95 [0,83-1,08]). Într-o analiză post-hoc a studiului 2 (pacienți cu alte tumori solide sau mielom multiplu), supraviețuirea globală a fost examinată pentru cele trei tipuri de tumori utilizate pentru stratificare (cancer pulmonar cu celule mici, mielom multiplu și multe altele). Supraviețuirea generală a fost mai mare pentru XGEVA în cancerul pulmonar cu celule mici (raport de risc [IC 95%] de 0,79 [0,65-0,95]; n = 702), mai mare pentru acidul zoledronic în mielom multiplu (raport de risc [IC 95%] de 2,26 [1,13- 4,50]; n = 180) și similar pentru XGEVA și acid zoledronic în alte tipuri de tumori (raport de pericol [IC 95%] de 1,08 [0,90-1,30]; n = 894). Factorii de prognostic și tratamentele antineoplazice nu au fost verificate în acest studiu. Într-o analiză combinată pre-specificată a studiilor 1, 2 și 3, supraviețuirea globală a fost similară între XGEVA și acid zoledronic (raport de pericol și IC 95%: 0,99 [0,91-1,07]).
Efecte asupra durerii
Timpul până la ameliorarea durerii (de exemplu, reducerea cu 2 puncte față de valoarea inițială, în cel mai scăzut scor de durere BPI-SF) a fost similar pentru denosumab și acid zoledronic în fiecare studiu și analize integrate. Într-o „analiză post-hoc a setului de date combinat, timpul mediu până la agravarea durerii (> 4 puncte în scorul de durere cu intensitatea cea mai slabă) la pacienții cu durere ușoară sau lipsă la momentul inițial a fost întârziat pentru XGEVA comparativ cu„ acidul zoledronic (198 vs. . 143 zile) (p = 0,0002).
Eficacitate clinică la adulți și adolescenți scheletici maturi cu tumoră osoasă cu celule uriașe
Siguranța și eficacitatea XGEVA au fost studiate în două studii clinice deschise, de fază II, cu un singur braț (studiile 4 și 5), în care au fost înscriși 529 pacienți cu tumori osoase de celule gigant, fie nerezecabile, fie nerezecabile. morbiditate.
Studiul 4 a înscris 37 de pacienți adulți cu tumoare osoasă cu celule uriașe nerezecabile confirmate histologic sau tumoare recidivantă cu celule uriașe osoase. Criteriile de răspuns au inclus eliminarea celulelor gigantice pe bază histopatologică sau absența progresiei pe bază radiografică.
Dintre cei 35 de pacienți incluși în analiza de eficacitate, 85,7% (IC 95%: 69,7-95,2) au avut un răspuns la tratamentul cu XGEVA. Toți cei 20 de pacienți (100%) care au fost supuși unei evaluări histologice au răspuns la tratament. La restul de 15 pacienți, 10 (67%) rapoarte radiologice nu au arătat o progresie a leziunii țintă.
Studiul 5 a înscris 507 adulți sau adolescenți maturi din punct de vedere scheletic cu tumori osoase cu celule uriașe și dovezi ale unei boli active măsurabile.
În Cohorta 1 (pacienți cu boală nerezecabilă), timpul mediu până la progresia bolii nu a fost atins, 21 din cei 258 de pacienți tratați aveau progresia bolii. În Cohorta 2 (pacienți cu boală rezecabilă, dar pentru care intervenția chirurgicală planificată a fost asociată cu morbiditate severă), 209 din 228 de pacienți evaluați tratați cu XGEVA nu au fost supuși intervenției chirurgicale în luna 6. În general, din 225 de pacienți pentru care a fost os cu tumoră celulară gigantă intervenția chirurgicală a fost programată (exclusiv metastazele pulmonare), 109 nu au suferit intervenții chirurgicale și 84 au suferit proceduri mai puțin invazive decât au fost planificate la momentul inițial. Timpul mediu până la intervenția chirurgicală a fost de 261 zile.
O revizuire retrospectivă independentă a datelor imagistice radiologice a fost efectuată la înscrierea a 305 pacienți în studiile 4 și 5. O sută nouăzeci au avut un răspuns evaluabil cel puțin o dată și au fost incluși în analiză (Tabelul 3). În general, XGEVA a obținut răspunsuri obiective la 71,6% dintre pacienți (95% CI: 64,6-77,9) (tabelul 3) evaluați utilizând diferite metodologii, majoritatea răspunsurilor fiind definite ca reducerea activității PET-ului fluorodeoxiglucozei sau creșterea densității măsurată în CT / HU, doar 25,1% dintre pacienți au avut un răspuns conform RECIST. Timpul mediu până la răspuns a fost de 3,1 luni (IÎ 95%: 2,89-3, 65) Durata medie a răspunsului nu a putut fi evaluată (patru pacienți au avut progresie a bolii în urma răspunsului obiectiv. ) La 190 de subiecți evaluabili pentru răspunsul obiectiv al tumorii, 55 de subiecți cu GCTB au fost supuși unei intervenții chirurgicale, dintre care 40 au suferit o rezecție completă.
Tabelul 3: Răspunsul obiectiv la tratament la pacienții cu cancer de oase cu celule uriașe
¹ CI = Interval de încredere exact
² RECIST 1.1: criterii modificate pentru evaluarea răspunsului în tumorile solide pentru a evalua masa tumorii prin intermediul tomografiei axiale computerizate (CT) sau a imaginii prin rezonanță magnetică (RMN).
³ EORTC: criterii modificate ale Organizației Europene pentru Cercetarea și Tratamentul Cancerului pentru a evalua răspunsul metabolic prin utilizarea tomografiei cu emisie de pozitroni cu fluorhidroxioglucoză (FDG-PET).
4 Densitate / dimensiune: Choi Criterii modificate invers pentru a evalua dimensiunea și densitatea tumorii folosind unități Hounsfield pe baza CT / RMN.
Efect asupra durerii
La înscrierea a 282 de pacienți, în studiul 5 cohorte combinate 1 și 2, a fost raportată o reducere semnificativă clinic a celei mai grave dureri (de exemplu, ≥ 2 puncte de scădere față de valoarea inițială) la 31,4% dintre pacienții cu risc (de exemplu, cei care au avut o durere mai gravă scorul de ≥ 2 la momentul inițial) în decurs de o săptămână de tratament și ≥ 50% la săptămâna 5. Aceste îmbunătățiri ale durerii au rămas neschimbate în evaluările ulterioare. , unde 74,8% dintre pacienți au raportat moderată sau neutilizarea analgezicelor (de exemplu, scor analgezic ≤ 2) și 25,2% dintre pacienți utilizează opioide puternice (de exemplu, scor analgezic 3-7).
Populația pediatrică
Agenția Europeană a Medicamentului a renunțat la obligația de a prezenta rezultatele studiilor cu XGEVA la toate subseturile populației pediatrice pentru prevenirea evenimentelor legate de schelet la pacienții cu metastaze osoase și la subseturile populației pediatrice sub 12 ani pentru tratamentul gigantului tumoră osoasă celulară (vezi pct. 4.2 pentru informații despre utilizarea la copii și adolescenți).
În studiul 5, XGEVA a fost evaluat într-un subgrup de 18 pacienți adolescenți (cu vârste cuprinse între 13 și 17 ani) cu tumoră osoasă cu celule gigantice care au atins maturitatea scheletului definită ca cel puțin un os lung matur (de exemplu, humerus cu creștere epifizară închis-închis pe disc plăcuță a humerusului) și greutatea corporală ≥ 45 kg. Un răspuns obiectiv a fost observat la patru din șase adolescenți evaluabili într-o analiză intermediară a studiului 5. O evaluare efectuată de un investigator a raportat că toți cei 18 pacienți adolescenți au avut cei mai buni stabili sau superiori răspunsul la boală (răspuns complet la 2 pacienți, răspuns parțial la 8 pacienți și stabilitatea bolii la 8 pacienți). Agenția Europeană pentru Medicamente a întârziat obligația de a prezenta rezultatele finale ale acestui studiu.
05.2 "Proprietăți farmacocinetice -
Absorbţie
După administrarea subcutanată, biodisponibilitatea a fost de 62%.
Biotransformare
Denosumab este compus exclusiv din aminoacizi și carbohidrați, cum ar fi imunoglobulinele native și este puțin probabil să fie eliminat prin mecanisme metabolice hepatice. Se așteaptă ca metabolismul și eliminarea medicamentelor să urmeze căile de eliminare a imunoglobulinei, adică degradarea în peptide mici și aminoacizi unici.
Eliminare
La subiecții cu cancer avansat, cărora li s-au administrat doze multiple de 120 mg la fiecare 4 săptămâni, s-a observat o acumulare de aproximativ 2 ori în concentrațiile serice de denosumab și s-a obținut starea de echilibru în decurs de 6 luni; acest lucru este în concordanță cu farmacocinetica independentă de timp. La subiecții cu tumoră osoasă cu celule gigantice care au primit 120 mg la fiecare 4 săptămâni, cu o doză de încărcare în zilele 8 și 15, nivelurile de echilibru au fost atinse în prima lună de tratament. Între săptămânile 9 și 49, nivelurile mediane au variat cu mai puțin de 9%. La subiecții care au încetat să mai ia 120 mg la fiecare 4 săptămâni, timpul de înjumătățire mediu a fost de 28 de zile (interval: 14-55 zile).
O analiză farmacocinetică a populației nu a relevat modificări semnificative clinic ale expunerii sistemice la denosumab la starea de echilibru pentru vârstă (18-87 ani), rasă / grup etnic (au fost studiați subiecții negri, hispanici, asiatici și albi.), Sexul sau tipul de tumoare solidă . Creșterea în greutate a fost asociată cu reduceri ale expunerii sistemice și invers. Modificările nu au fost considerate relevante din punct de vedere clinic, deoarece efectele farmacodinamice bazate pe markerii de rotație osoasă au fost constante pe o gamă largă de greutăți corporale.
Liniaritate / neliniaritate
Denosumab a prezentat farmacocinetică neliniară la diferite niveluri de doză, dar pentru doze de 60 mg (sau 1 mg / kg) și peste a prezentat creșteri ale dozei aproximativ proporționale cu expunerea. Non-liniaritatea este probabil datorită unui mecanism de eliminare. important la concentrații scăzute.
Insuficiență renală
În studiile cu denosumab (60 mg, n = 55 și 120 mg, n = 32) la pacienții fără cancer avansat, dar cu grade variate ale funcției renale, inclusiv pacienții dializați, gradul de insuficiență renală nu a avut niciun efect asupra farmacocineticii denosumab; de aceea nu este necesară ajustarea dozei în caz de insuficiență renală. Monitorizarea renală nu este necesară la administrarea XGEVA.
Insuficiență hepatică
Nu s-au efectuat studii specifice la pacienții cu insuficiență hepatică. În general, anticorpii monoclonali nu sunt eliminați prin metabolismul hepatic. Se așteaptă ca farmacocinetica Denosumab să nu fie afectată de afectarea funcției hepatice.
Persoane în vârstă
În general, nu s-au observat diferențe în ceea ce privește siguranța și eficacitatea între pacienții geriatrici și pacienții mai tineri. este necesară la pacienții vârstnici.
Populația pediatrică
Profilul farmacocinetic la populația pediatrică nu a fost evaluat.
05.3 Date preclinice de siguranță -
Deoarece activitatea biologică a denosumabului la animale este specifică primatelor neumane, evaluările șoarecilor modificați genetic (knockout) sau utilizarea altor inhibitori biologici ai căii au fost utilizate pentru a evalua proprietățile farmacodinamice ale denosumab în modelele de rozătoare. RANK / RANKL , cum ar fi OPG-Fc și RANK-Fc.
La modelele de șoarece de metastaze osoase ale cancerului de sân uman, receptor de estrogen pozitiv și negativ, cancer de prostată și cancer pulmonar cu celule mici, OPG-Fc a redus leziunile osteolitice, osteoblastice și osteolitice / osteoblastice, a întârziat formarea metastazelor osoase de novo și reducerea creșterii tumorale a sistemului osos. În aceste modele, când OPG-Fc a fost combinat cu terapia hormonală (tamoxifen) sau chimioterapie (docetaxel), s-a găsit o „inhibare suplimentară a creșterii tumorale a sistemului osos. în cancerul de sân și prostată sau respectiv cancer pulmonar. Într-un model de șoarece de inducere a cancerului de sân, RANK-Fc a redus proliferarea indusă de hormoni în epiteliul mamar și a întârziat formarea tumorii.
Nu s-au efectuat teste standard pentru investigarea potențialei genotoxicități a denosumabului, deoarece aceste teste nu sunt relevante pentru această moleculă. Cu toate acestea, având în vedere caracteristicile sale, este puțin probabil ca denosumab să aibă potențial genotoxic.
Potențialul cancerigen al denosumab nu a fost evaluat în studii pe termen lung pe animale.
În studiile de toxicitate cu doză unică și repetate efectuate la maimuțele cynomolgus, dozele de denosumab care au dus la „expunere sistemică de 2,7 până la 15 ori doza recomandată la om nu au avut niciun impact asupra fiziologiei cardiovasculare, asupra fertilității masculine sau asupra femelei sau asupra produsului specific pentru toxicitatea organelor.
Într-un studiu efectuat pe maimuțe cynomolgus cu doze multiple de denosumab în perioada echivalentă cu primul trimestru de sarcină, dozele de denosumab care au dus la o expunere sistemică de 9 ori a dozei recomandate la om nu au indus toxicitate maternă sau vătămare a fătului o perioadă echivalentă cu primul trimestru; totuși, ganglionii limfatici fetali nu au fost examinați.
Într-un alt studiu al maimuțelor cynomolgus cărora li s-a administrat denosumab în timpul sarcinii, la o expunere sistemică de 12 ori mai mare decât doza umană, a existat o creștere a mortalității și a mortalității postnatale; absența ganglionilor limfatici periferici; și creșterea neonatală redusă. Nu a fost stabilit un nivel de doză care poate fi negativ pentru efectele asupra reproducerii. Ulterior, la 6 luni după naștere, modificările osoase au arătat o recuperare și nu a existat niciun efect asupra erupției dentare. Cu toate acestea, efectele asupra ganglionilor limfatici și malaliniere dentară au persistat și a fost observată într-o mineralizare de la minim la moderat în mai multe țesuturi (tratament corelație incertă) .Nu au existat dovezi ale afectării materne înainte de travaliu. Efectele adverse materne au apărut rar în timpul travaliului. Dezvoltarea glandei mamare materne a fost normală.
În studiile preclinice privind calitatea osoasă efectuate pe maimuțe tratate pe termen lung cu denosumab, scăderea fluctuației osoase a fost însoțită de o rezistență osoasă îmbunătățită și histologie normală.
La șoarecii masculi proiectați genetic pentru a exprima RANKL uman (șoareci knock-in) și supuși unei fracturi transcorticale, denosumab a întârziat îndepărtarea cartilajului și remodelarea calului în comparație cu grupul martor, dar puterea biomecanică nu a fost afectată negativ.
Absența alăptării datorită inhibării maturării glandei mamare (dezvoltarea structurilor lobulo-alveolare ale glandei mamare în timpul sarcinii) a fost observată la șoarecii knockout care nu au exprimat RANK sau RANKL, precum și formarea afectată Șoarecii nou-născuți knockout RANK / RANKL pierderea în greutate, scăderea creșterii osoase, modificarea plăcilor de creștere și lipsa erupției dentare. Creșterea osoasă redusă, modificarea plăcilor de creștere și alterarea erupției dentare. au fost observate și în studiile la șobolani neonatali cărora li s-au administrat inhibitori RANKL; aceste modificări au fost parțial reversibile după întreruperea inhibitorului RANKL Primate adolescente tratate cu doze de denosumab de 2,7 și 15 ori (doze de 10 și 50 mg / kg) au prezentat anomalii ale liniile de creștere. Prin urmare, tratamentul cu denosumab poate afecta creșterea osoasă la copiii cu plăci de creștere deschise și poate inhiba erupția dentară.
06.0 INFORMAȚII FARMACEUTICE -
06.1 Excipienți -
Acid acetic glacial *
Hidroxid de sodiu (pentru ajustarea pH-ului) *
Sorbitol (E420)
Apă pentru preparate injectabile
* Tamponul acetat se obține prin amestecarea acidului acetic și a hidroxidului de sodiu
06.2 Incompatibilitate "-
În absența studiilor de compatibilitate, acest medicament nu trebuie amestecat cu alte medicamente.
06.3 Perioada de valabilitate "-
3 ani.
XGEVA poate fi păstrat la temperatura camerei (până la 25 ° C) timp de până la 30 de zile în ambalajul original. Odată scos din frigider, XGEVA trebuie utilizat în această perioadă de 30 de zile.
06.4 Precauții speciale pentru depozitare -
A se păstra la frigider (2 ° C - 8 ° C).
Nu înghețați.
Păstrați flaconul în cutie pentru a proteja medicamentul de lumină.
06.5 Natura ambalajului imediat și conținutul ambalajului -
Soluție de 1,7 ml într-un flacon de unică folosință (sticlă tip I) cu dop (acoperit cu fluoropolimer elastomer) și o garnitură (aluminiu) cu un capac rabatabil.
Dimensiunea ambalajului de una, trei sau patru fiole.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
06.6 Instrucțiuni de utilizare și manipulare -
Înainte de administrare, soluția XGEVA trebuie inspectată vizual. Soluția poate conține urme de particule de proteine translucide până la albe.Nu injectați soluția dacă este tulbure sau decolorată Nu agitați excesiv Pentru a evita problemele de la locul injectării, permiteți flaconului să atingă temperatura camerei (până la 25 ° C) înainte de injectare și injectați încet. Injectați întregul conținut al flaconului. Pentru administrarea denosumab se recomandă un ac de oțel cu calibru 27. Nu refolosiți flaconul.
Medicamentele neutilizate și deșeurile derivate din acest medicament trebuie eliminate în conformitate cu reglementările locale.
07.0 DEȚINĂTORUL „AUTORIZAȚIEI DE PUNERE PE PIAȚĂ” -
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
Olanda
08.0 NUMĂRUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ -
EU / 1/11/703/001
EU / 1/11/703/002
EU / 1/11/703/003
041300017
041300029
09.0 DATA PRIMEI AUTORIZAȚII SAU REÎNNOIREA AUTORIZAȚIEI -
Data primei autorizații: 13 iulie 2011
Data ultimei reînnoiri: 4 aprilie 2016
10.0 DATA REVIZUIRII TEXTULUI -
Decembrie 2016















-nelle-carni-di-maiale.jpg)